Combined dataset
Zoom in a specific population
2025-05-03
Source:vignettes/examples/2_combined/2_subdataset.Rmd
2_subdataset.RmdThis notebook shows the use of these functions from
aquarius:
aquarius::filter_featuresaquarius::integration_fastmnnaquarius::run_diffusion_mapaquarius::plot_dfaquarius::palette_GrOrBl
The goal of this script is to merge the two filtered individual datasets, called A and B, by considering only a specific cell population.
## [1] "/usr/lib/R/library" "/usr/local/lib/R/site-library"
## [3] "/usr/lib/R/site-library"Preparation
In this section, we set the global settings of the analysis. We will store data there:
out_dir = "."The dataset will have this name:
save_name = "tumor_cells"The filtered Seurat objects are stored there:
data_path = paste0(out_dir, "/../1_individual/datasets/")
list.files(data_path)## [1] "A_sobj_filtered.rds" "A_sobj_unfiltered.rds" "B_sobj_filtered.rds"
## [4] "B_sobj_unfiltered.rds"Here are custom colors for each cell type:
color_markers = c("macrophages" = "#6ECEDF",
"tumor cells" = "#DA2328")We define custom colors for each sample:
sample_info = data.frame(
project_name = c("A", "B"),
sample_identifier = c("A", "B"),
color = c("#7B52AE", "#74B652"),
row.names = c("A", "B"))
aquarius::plot_df(sample_info)
We set main parameters:
n_threads = 10L # Rtsne::Rtsne optionMake tumor_cells dataset
Individual datasets
For each sample, we:
- load individual dataset
- look at cell annotation
We load individual datasets:
sobj_list = lapply(sample_info$project_name, FUN = function(one_project_name) {
sobj = readRDS(paste0(data_path, one_project_name, "_sobj_filtered.rds"))
return(sobj)
})
names(sobj_list) = sample_info$project_name
lapply(sobj_list, FUN = dim) %>%
do.call(rbind, .) %>%
rbind(., colSums(.)) %>%
`colnames<-`(c("Nb genes", "Nb cells"))## Nb genes Nb cells
## A 28000 563
## B 28000 545
## 56000 1108We represent cells in the tSNE:
name2D = "RNA_pca_20_tsne"We are interested by this population:
cell_type_of_interest = c("tumor cells")Extract cells of interest
One may choose one method among three possible:
- single-cell level annotation: suited when the cluster annotation does not present satisfying results. But, cells suffering from annotation competition may be mis-selected (false positives or false negatives)
- smoothed cluster annotation: a way to automate the selection of clusters without giving the explicit clusters identities. It smooths the single-cell level annotation at a cluster-level, i.e., clusters are annotated for the main cell type it contains.
- average gene expression levels: to select several cell types, for instance several immune cell types, you can set a vector containing the cell types of interest (methods 1 and 2), or define several markers of interest (eg. Cd45 / Ptprc). The cells will be scored for the expression levels of markers of interest. Then, you can select the cells having a score greater than a threshold or, alternatively, the clusters having an average score greater than a threshold.
Negative selection can be also considered, i.e. filtering cells that do not correspond to an annotation/score.
Method 1: single-cell level annotation
We look at cell type annotation for each dataset:
plot_list = lapply(sobj_list, FUN = function(sobj) {
mytitle = as.character(unique(sobj@project.name))
mysubtitle = ncol(sobj)
p = Seurat::DimPlot(sobj, group.by = "cell_type",
reduction = name2D) +
ggplot2::scale_color_manual(values = color_markers,
breaks = names(color_markers),
name = "Cell Type") +
ggplot2::labs(title = mytitle,
subtitle = paste0(mysubtitle, " cells")) +
ggplot2::theme(aspect.ratio = 1,
plot.title = element_text(hjust = 0.5),
plot.subtitle = element_text(hjust = 0.5)) +
Seurat::NoAxes()
return(p)
})
plot_list[[length(plot_list) + 1]] = patchwork::guide_area()
patchwork::wrap_plots(plot_list, nrow = 1) +
patchwork::plot_layout(guides = "collect") &
ggplot2::theme(legend.position = "right")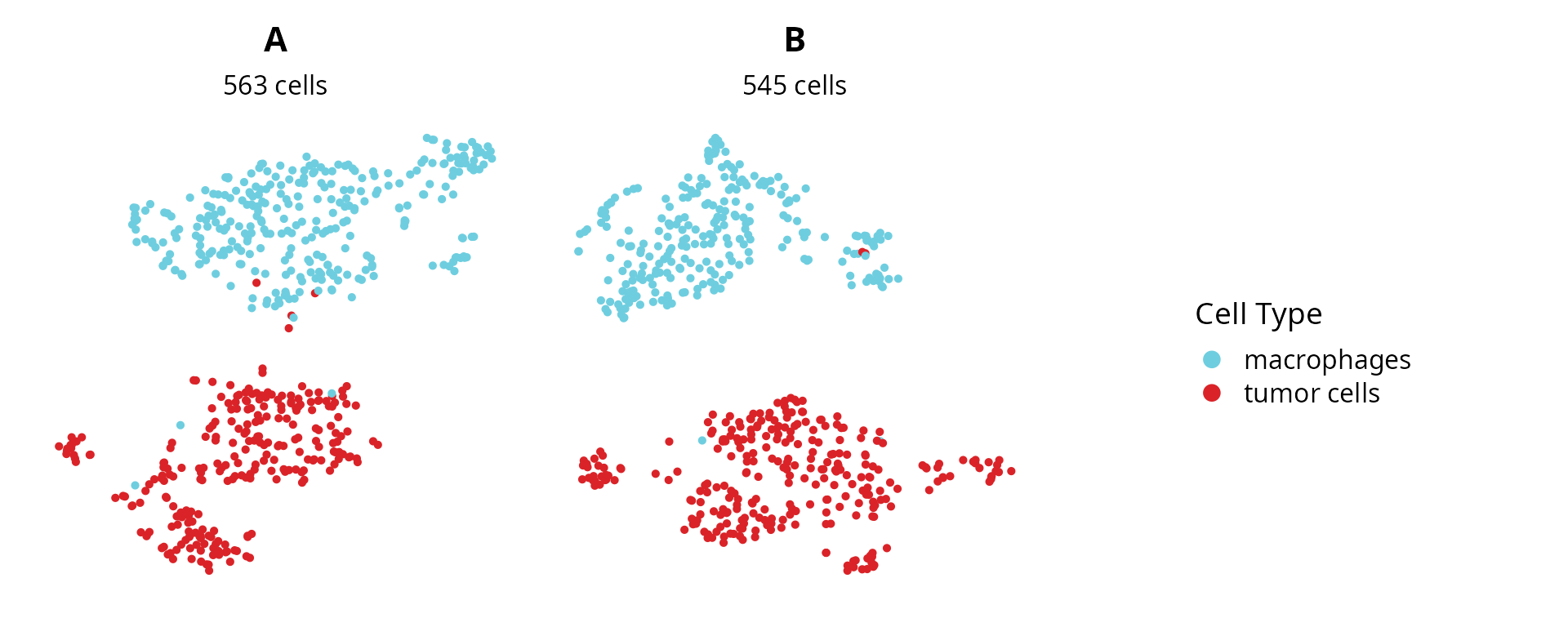
We annotate the cells to keep:
sobj_list = lapply(sobj_list, FUN = function(one_sobj) {
one_sobj$is_of_interest = (one_sobj$cell_type %in% cell_type_of_interest)
return(one_sobj)
})
lapply(sobj_list, FUN = function(one_sobj) {
return(table(one_sobj$is_of_interest))
}) %>% do.call(rbind, .) %>%
rbind(., colSums(.))## FALSE TRUE
## A 308 255
## B 261 284
## 569 539Method 2: smoothed cluster annotation
We smooth cell type annotation at a cluster level:
sobj_list = lapply(sobj_list, FUN = function(one_sobj) {
cluster_type = table(one_sobj$cell_type, one_sobj$seurat_clusters) %>%
prop.table(., margin = 2) %>%
apply(., 2, which.max)
cluster_type = setNames(nm = names(cluster_type),
levels(one_sobj$cell_type)[cluster_type])
one_sobj$cluster_type = setNames(nm = names(one_sobj$seurat_clusters),
cluster_type[one_sobj$seurat_clusters])
## Output
return(one_sobj)
})We visualize the score for the cell type of interest, the cell type annotation, the clustering, and the cluster annotation.
plot_list = lapply(sobj_list, FUN = function(one_sobj) {
project_name = as.character(unique(one_sobj$orig.ident))
plot_sublist = list()
# Score
the_score = paste0("score_", gsub(x = cell_type_of_interest,
pattern = " ",
replacement = "_"))
plot_sublist[[1]] = Seurat::FeaturePlot(one_sobj, reduction = name2D,
features = the_score) +
ggplot2::labs(title = project_name,
subtitle = the_score) +
Seurat::NoAxes() +
ggplot2::scale_color_gradientn(colors = aquarius:::palette_GrOrBl) +
ggplot2::theme(aspect.ratio = 1,
plot.subtitle = element_text(hjust = 0.5))
# Cell type
plot_sublist[[2]] = Seurat::DimPlot(one_sobj,
reduction = name2D,
group.by = "cell_type",
cols = color_markers) +
ggplot2::labs(title = "Cell type annotation",
subtitle = paste0(sum(one_sobj$cell_type %in% cell_type_of_interest),
" cells")) +
Seurat::NoAxes() + Seurat::NoLegend() +
ggplot2::theme(aspect.ratio = 1,
plot.title = element_text(hjust = 0.5),
plot.subtitle = element_text(hjust = 0.5))
# Clusters
plot_sublist[[3]] = Seurat::DimPlot(one_sobj,
reduction = name2D,
group.by = "seurat_clusters",
label = TRUE) +
ggplot2::labs(title = "Clusters") +
Seurat::NoAxes() + Seurat::NoLegend() +
ggplot2::theme(aspect.ratio = 1,
plot.title = element_text(hjust = 0.5),
plot.subtitle = element_text(hjust = 0.5))
# Cluster type
plot_sublist[[4]] = Seurat::DimPlot(one_sobj,
reduction = name2D,
group.by = "cluster_type",
cols = color_markers) +
ggplot2::labs(title = "Cluster annotation",
subtitle = paste0(sum(one_sobj$cluster_type %in% cell_type_of_interest),
" cells")) +
Seurat::NoAxes() + Seurat::NoLegend() +
ggplot2::theme(aspect.ratio = 1,
plot.title = element_text(hjust = 0.5),
plot.subtitle = element_text(hjust = 0.5))
return(plot_sublist)
}) %>% unlist(., recursive = FALSE)
patchwork::wrap_plots(plot_list, ncol = 4)
We annotate the cells to keep:
sobj_list = lapply(sobj_list, FUN = function(one_sobj) {
one_sobj$is_of_interest = (one_sobj$cluster_type %in% cell_type_of_interest)
return(one_sobj)
})
lapply(sobj_list, FUN = function(one_sobj) {
return(table(one_sobj$is_of_interest))
}) %>% do.call(rbind, .) %>%
rbind(., colSums(.))## FALSE TRUE
## A 310 253
## B 261 284
## 571 537Method 3: average gene expression levels
We define a set of markers of interest. Here, we provide the exact same genes used for the cell type annotation. But, custom gene sets can be provided.
markers_of_interest = c("Sox9", "Sox10", "Ank3", "Plp1", "Col16a1")We attribute a score for each cell:
sobj_list = lapply(sobj_list, FUN = function(one_sobj) {
one_sobj$score_of_interest = Seurat::AddModuleScore(one_sobj,
features = list(markers_of_interest))$Cluster1
return(one_sobj)
})We set an arbitrary threshold for the score:
score_thresh = 0.2We visualize the score, the cells with a score above the threshold, the clustering, and the average score by cluster.
plot_list = lapply(sobj_list, FUN = function(one_sobj) {
project_name = as.character(unique(one_sobj$orig.ident))
plot_sublist = list()
# Score
plot_sublist[[1]] = Seurat::FeaturePlot(one_sobj, reduction = name2D,
features = "score_of_interest") +
ggplot2::labs(title = project_name,
subtitle = "Score for genes of interest") +
Seurat::NoAxes() +
ggplot2::scale_color_gradientn(colors = aquarius:::palette_GrOrBl) +
ggplot2::theme(aspect.ratio = 1,
plot.subtitle = element_text(hjust = 0.5))
# Cells with high score
one_sobj$score_above_threshold = (one_sobj$score_of_interest > score_thresh)
plot_sublist[[2]] = Seurat::DimPlot(one_sobj,
reduction = name2D,
group.by = "score_above_threshold") +
ggplot2::labs(title = paste0("Score thresold: ", score_thresh),
subtitle = paste0(sum(one_sobj$score_above_threshold), " cells")) +
Seurat::NoAxes() + Seurat::NoLegend() +
ggplot2::theme(aspect.ratio = 1,
plot.title = element_text(hjust = 0.5),
plot.subtitle = element_text(hjust = 0.5))
# Clusters
plot_sublist[[3]] = Seurat::DimPlot(one_sobj,
reduction = name2D,
group.by = "seurat_clusters",
label = TRUE) +
ggplot2::labs(title = "Clusters") +
Seurat::NoAxes() + Seurat::NoLegend() +
ggplot2::theme(aspect.ratio = 1,
plot.title = element_text(hjust = 0.5),
plot.subtitle = element_text(hjust = 0.5))
# Score by cluster
score_by_clusters = one_sobj@meta.data %>%
dplyr::select(seurat_clusters, score_of_interest) %>%
dplyr::group_by(seurat_clusters) %>%
dplyr::summarise(avg_score = mean(score_of_interest)) %>%
as.data.frame()
clusters_high_score = score_by_clusters %>%
dplyr::filter(avg_score > score_thresh) %>%
dplyr::pull(seurat_clusters)
nb_cells_in_clusters = sum(one_sobj$seurat_clusters %in% clusters_high_score)
plot_sublist[[4]] = ggplot2::ggplot(score_by_clusters, aes(x = seurat_clusters, y = avg_score)) +
ggplot2::geom_point() +
ggplot2::geom_hline(yintercept = score_thresh, col = "red") +
ggplot2::labs(x = "Cluster ID", y = "Mean score",
title = paste0("Score thresold: ", score_thresh),
subtitle = paste0(nb_cells_in_clusters, " cells")) +
ggplot2::theme_classic() +
ggplot2::theme(plot.title = element_text(hjust = 0.5, face = "bold"),
plot.subtitle = element_text(hjust = 0.5))
return(plot_sublist)
}) %>% unlist(., recursive = FALSE)
patchwork::wrap_plots(plot_list, ncol = 4)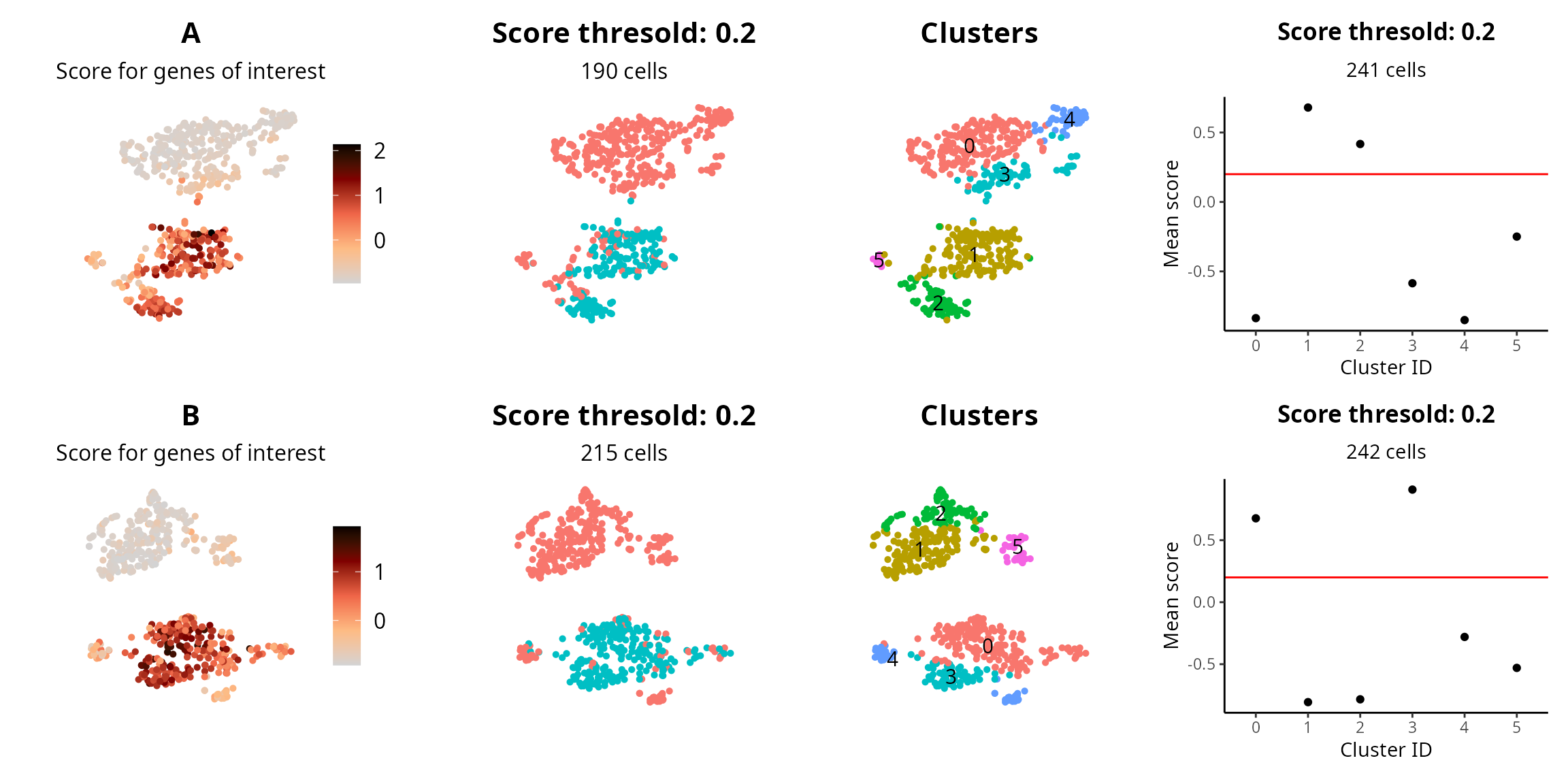
We annotate the cells to keep, based on the single-cell level score:
sobj_list = lapply(sobj_list, FUN = function(one_sobj) {
one_sobj$is_of_interest = (one_sobj$score_of_interest > score_thresh)
return(one_sobj)
})
lapply(sobj_list, FUN = function(one_sobj) {
return(table(one_sobj$is_of_interest))
}) %>% do.call(rbind, .) %>%
rbind(., colSums(.))## FALSE TRUE
## A 373 190
## B 330 215
## 703 405Alternatively, based on the average score by cluster:
sobj_list = lapply(sobj_list, FUN = function(one_sobj) {
clusters_high_score = one_sobj@meta.data %>%
dplyr::select(seurat_clusters, score_of_interest) %>%
dplyr::group_by(seurat_clusters) %>%
dplyr::summarise(avg_score = mean(score_of_interest)) %>%
dplyr::filter(avg_score > score_thresh) %>%
dplyr::pull(seurat_clusters)
one_sobj$is_of_interest = (one_sobj$seurat_clusters %in% clusters_high_score)
return(one_sobj)
})
lapply(sobj_list, FUN = function(one_sobj) {
return(table(one_sobj$is_of_interest))
}) %>% do.call(rbind, .) %>%
rbind(., colSums(.))## FALSE TRUE
## A 322 241
## B 303 242
## 625 483Combined dataset
For this example, we extract cells based on the cluster-smoothed cell type annotation.
sobj_list = lapply(sobj_list, FUN = function(one_sobj) {
one_sobj$is_of_interest = (one_sobj$cluster_type %in% cell_type_of_interest)
return(one_sobj)
})
lapply(sobj_list, FUN = function(one_sobj) {
return(table(one_sobj$is_of_interest))
}) %>% do.call(rbind, .) %>%
rbind(., colSums(.))## FALSE TRUE
## A 310 253
## B 261 284
## 571 537We subset the individual objects:
sobj_list = lapply(sobj_list, FUN = function(one_sobj) {
if (sum(one_sobj$is_of_interest) > 0) {
one_sobj = subset(one_sobj, is_of_interest == TRUE)
} else {
one_sobj = NA
}
return(one_sobj)
})
lapply(sobj_list, FUN = dim) %>%
do.call(rbind, .) %>%
rbind(., colSums(.)) %>%
`colnames<-`(c("Nb genes", "Nb cells"))## Nb genes Nb cells
## A 28000 253
## B 28000 284
## 56000 537We combine the dataset:
sobj = base::merge(sobj_list[[1]],
y = sobj_list[c(2:length(sobj_list))],
add.cell.ids = names(sobj_list))
sobj## An object of class Seurat
## 28000 features across 537 samples within 1 assay
## Active assay: RNA (28000 features, 3000 variable features)
## 6 layers present: counts.A, counts.B, data.A, scale.data.A, data.B, scale.data.BWe merge the layers:
sobj = SeuratObject::JoinLayers(sobj, layers = "counts")
sobj = SeuratObject::JoinLayers(sobj, layers = "data")
sobj = SeuratObject::JoinLayers(sobj, layers = "scale.data")
sobj## An object of class Seurat
## 28000 features across 537 samples within 1 assay
## Active assay: RNA (28000 features, 3000 variable features)
## 3 layers present: scale.data, data, countsWe add again the correspondence between gene names and gene ID. Since all datasets have been aligned using the same transcriptome, we take the correspondence from one individual dataset.
sobj@assays$RNA@meta.data = sobj_list[[1]]@assays$RNA@meta.data[, c("Ensembl_ID", "gene_name")]
head(sobj@assays$RNA@meta.data)## Ensembl_ID gene_name
## Xkr4 ENSMUSG00000051951 Xkr4
## Gm1992 ENSMUSG00000089699 Gm1992
## Gm37381 ENSMUSG00000102343 Gm37381
## Rp1 ENSMUSG00000025900 Rp1
## Rp1.1 ENSMUSG00000109048 Rp1
## Sox17 ENSMUSG00000025902 Sox17We remove the list of objects:
rm(sobj_list)What is are the cells metadata ?
sobj$is_of_interest = NULL
sobj$score_of_interest = NULL
summary(sobj@meta.data)## orig.ident nCount_RNA nFeature_RNA log_nCount_RNA
## Length:537 Min. : 1022 Min. : 601 Min. : 6.930
## Class :character 1st Qu.: 2349 1st Qu.:1056 1st Qu.: 7.762
## Mode :character Median : 3878 Median :1667 Median : 8.263
## Mean : 5792 Mean :2011 Mean : 8.351
## 3rd Qu.: 6666 3rd Qu.:2553 3rd Qu.: 8.805
## Max. :33763 Max. :6042 Max. :10.427
## Seurat.Phase cyclone.Phase RNA_snn_res.0.5 seurat_clusters
## Length:537 Length:537 Length:537 Length:537
## Class :character Class :character Class :character Class :character
## Mode :character Mode :character Mode :character Mode :character
##
##
##
## percent.mt percent.rb score_macrophages score_tumor_cells
## Min. : 0.000 Min. : 0.1894 Min. :-1.2490 Min. :-0.7305
## 1st Qu.: 5.643 1st Qu.: 1.1910 1st Qu.:-1.0310 1st Qu.: 0.1883
## Median :10.242 Median : 3.0620 Median :-0.8385 Median : 0.6353
## Mean :10.559 Mean : 4.8629 Mean :-0.8324 Mean : 0.5720
## 3rd Qu.:15.902 3rd Qu.: 7.2975 3rd Qu.:-0.6642 3rd Qu.: 0.9543
## Max. :19.959 Max. :19.9397 Max. : 0.9580 Max. : 2.2151
## cell_type cluster_type
## Length:537 Length:537
## Class :character Class :character
## Mode :character Mode :character
##
##
## Processing
We remove genes that are expressed in less than 5 cells:
sobj = aquarius::filter_features(sobj, min_cells = 5)
sobj## An object of class Seurat
## 12361 features across 537 samples within 1 assay
## Active assay: RNA (12361 features, 0 variable features)
## 3 layers present: scale.data, data, countsProjection
We normalize the count matrix for remaining cells and select highly variable features:
sobj = Seurat::NormalizeData(sobj,
normalization.method = "LogNormalize")
sobj = Seurat::FindVariableFeatures(sobj, nfeatures = 2000)
sobj = Seurat::ScaleData(sobj)
sobj## An object of class Seurat
## 12361 features across 537 samples within 1 assay
## Active assay: RNA (12361 features, 2000 variable features)
## 3 layers present: scale.data, data, countsWe perform a PCA:
sobj = Seurat::RunPCA(sobj,
assay = "RNA",
reduction.name = "RNA_pca",
npcs = 100,
seed.use = 1337L)
sobj## An object of class Seurat
## 12361 features across 537 samples within 1 assay
## Active assay: RNA (12361 features, 2000 variable features)
## 3 layers present: scale.data, data, counts
## 1 dimensional reduction calculated: RNA_pcaWe choose the number of dimensions such that they summarize 60 % of the variability:
stdev = sobj@reductions[["RNA_pca"]]@stdev
stdev_prop = cumsum(stdev)/sum(stdev)
ndims = which(stdev_prop > 0.60)[1]
ndims## [1] 51We can visualize this on the elbow plot:
elbow_p = Seurat::ElbowPlot(sobj, ndims = 100, reduction = "RNA_pca") +
ggplot2::geom_point(x = ndims, y = stdev[ndims], col = "red")
x_text = ggplot_build(elbow_p)$layout$panel_params[[1]]$x$get_labels() %>% as.numeric()
elbow_p = elbow_p +
ggplot2::scale_x_continuous(breaks = sort(c(x_text, ndims)), limits = c(0, 100))
x_color = ifelse(ggplot_build(elbow_p)$layout$panel_params[[1]]$x$get_labels() %>%
as.numeric() %>% round(., 2) == round(ndims, 2), "red", "black")
elbow_p = elbow_p +
ggplot2::theme_classic() +
ggplot2::theme(axis.text.x = element_text(color = x_color))
elbow_p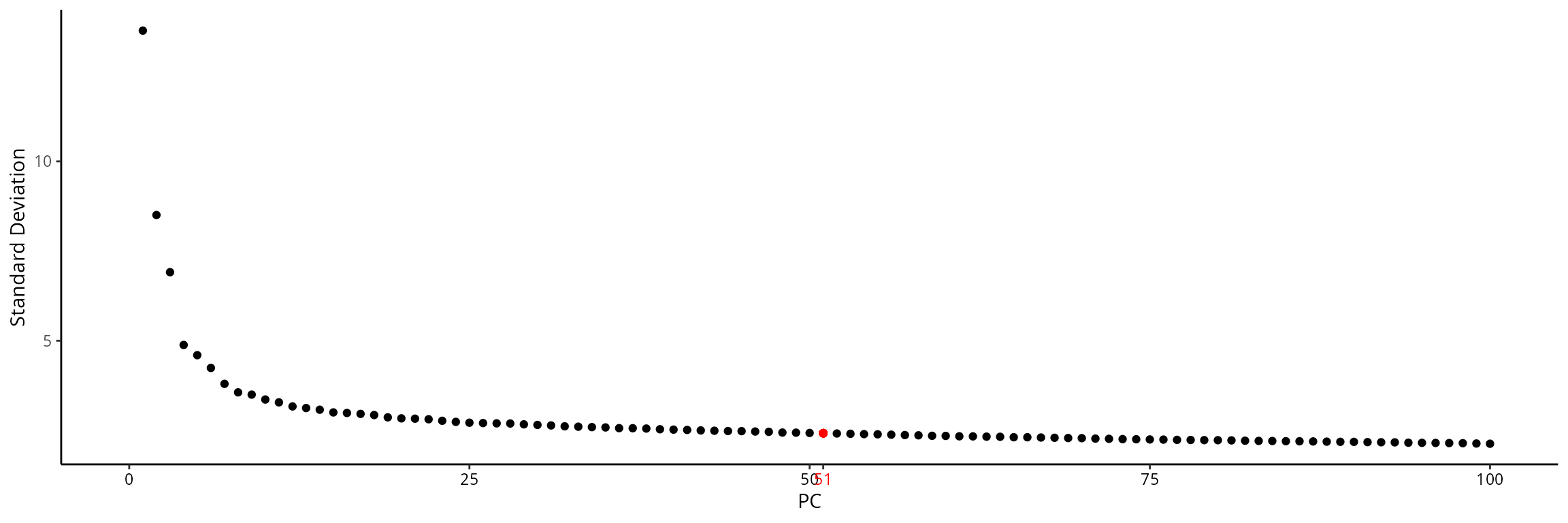
We generate a tSNE and a UMAP with 51 principal components:
sobj = Seurat::RunTSNE(sobj,
reduction = "RNA_pca",
dims = 1:ndims,
seed.use = 1337L,
num_threads = n_threads, # Rtsne::Rtsne option
reduction.name = paste0("RNA_pca_", ndims, "_tsne"))
sobj = Seurat::RunUMAP(sobj,
reduction = "RNA_pca",
dims = 1:ndims,
seed.use = 1337L,
reduction.name = paste0("RNA_pca_", ndims, "_umap"))(Time to run: 3.74 s)
We can visualize the two representations:
tsne = Seurat::DimPlot(sobj, group.by = "orig.ident",
reduction = paste0("RNA_pca_", ndims, "_tsne")) +
ggplot2::scale_color_manual(values = sample_info$color,
breaks = sample_info$project_name) +
Seurat::NoAxes() + ggplot2::ggtitle("PCA - tSNE") +
ggplot2::theme(aspect.ratio = 1,
plot.title = element_text(hjust = 0.5),
legend.position = "none")
umap = Seurat::DimPlot(sobj, group.by = "orig.ident",
reduction = paste0("RNA_pca_", ndims, "_umap")) +
ggplot2::scale_color_manual(values = sample_info$color,
breaks = sample_info$project_name) +
Seurat::NoAxes() + ggplot2::ggtitle("PCA - UMAP") +
ggplot2::theme(aspect.ratio = 1,
plot.title = element_text(hjust = 0.5))
tsne | umap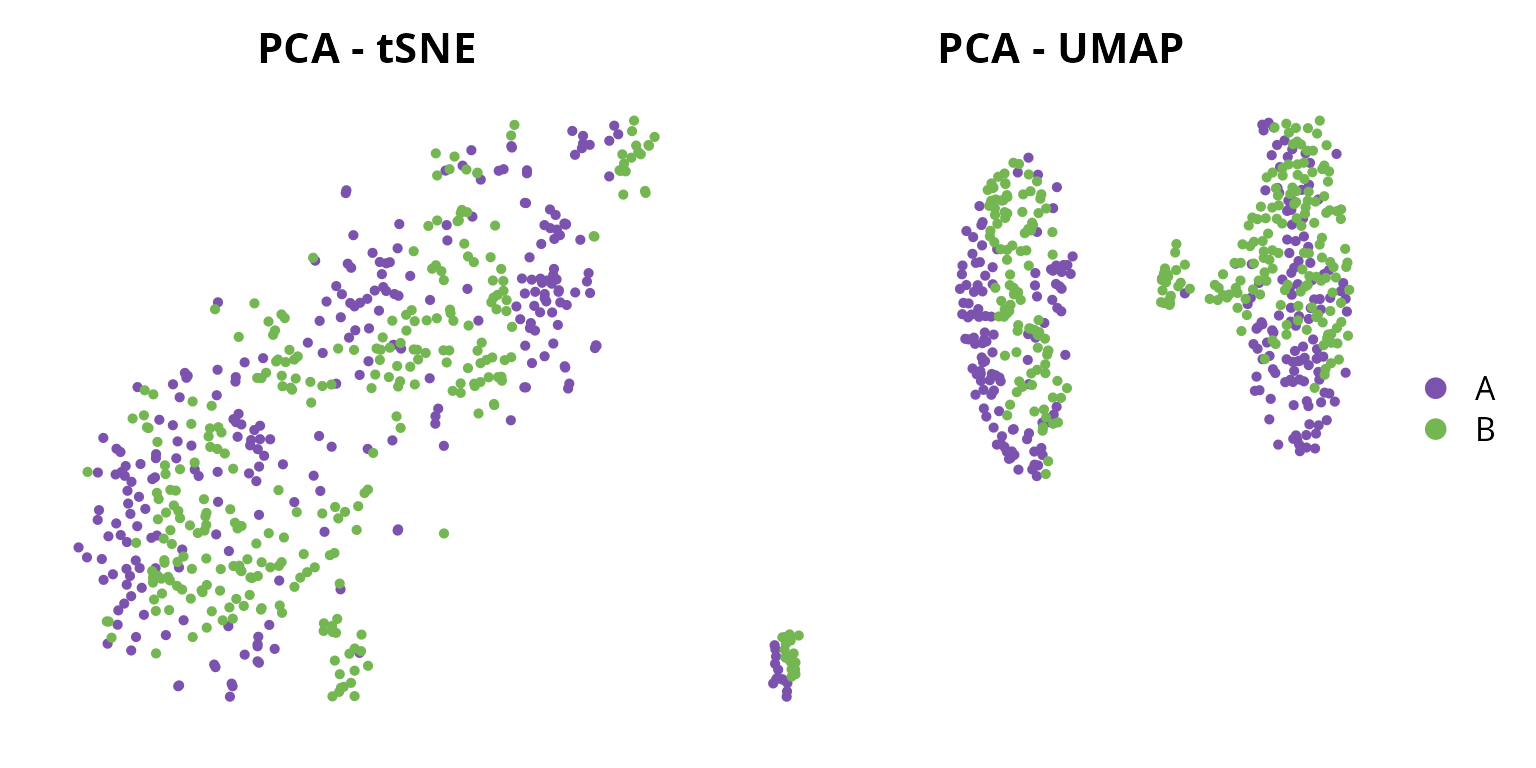
Batch-effect correction
We remove sample specific effect on the pca using
FastMNN:
sobj = aquarius::integration_fastmnn(sobj = sobj,
split_by = "orig.ident",
assay = "RNA",
verbose = TRUE,
d = 100)
sobj## An object of class Seurat
## 14361 features across 537 samples within 2 assays
## Active assay: RNA (12361 features, 2000 variable features)
## 3 layers present: scale.data, data, counts
## 1 other assay present: mnn.reconstructed
## 4 dimensional reductions calculated: RNA_pca, RNA_pca_51_tsne, RNA_pca_51_umap, mnn(Time to run: 6.89 s)
From this batch-effect removed projection, we generate a tSNE and a UMAP.
sobj = Seurat::RunUMAP(sobj,
seed.use = 1337L,
dims = 1:ndims,
reduction = "mnn",
reduction.name = paste0("mnn_", ndims, "_umap"),
reduction.key = paste0("mnn_", ndims, "umap_"))
sobj = Seurat::RunTSNE(sobj,
dims = 1:ndims,
seed.use = 1337L,
num_threads = n_threads, # Rtsne::Rtsne option
reduction = "mnn",
reduction.name = paste0("mnn_", ndims, "_tsne"),
reduction.key = paste0("mnn", ndims, "tsne_"))(Time to run: 3.88 s)
We visualize the corrected projections:
tsne = Seurat::DimPlot(sobj, group.by = "orig.ident",
reduction = paste0("mnn_", ndims, "_tsne")) +
ggplot2::scale_color_manual(values = sample_info$color,
breaks = sample_info$project_name) +
Seurat::NoAxes() + ggplot2::ggtitle("PCA - mnn - tSNE") +
ggplot2::theme(aspect.ratio = 1,
plot.title = element_text(hjust = 0.5),
legend.position = "none")
umap = Seurat::DimPlot(sobj, group.by = "orig.ident",
reduction = paste0("mnn_", ndims, "_umap")) +
ggplot2::scale_color_manual(values = sample_info$color,
breaks = sample_info$project_name) +
Seurat::NoAxes() + ggplot2::ggtitle("PCA - mnn - UMAP") +
ggplot2::theme(aspect.ratio = 1,
plot.title = element_text(hjust = 0.5))
tsne | umap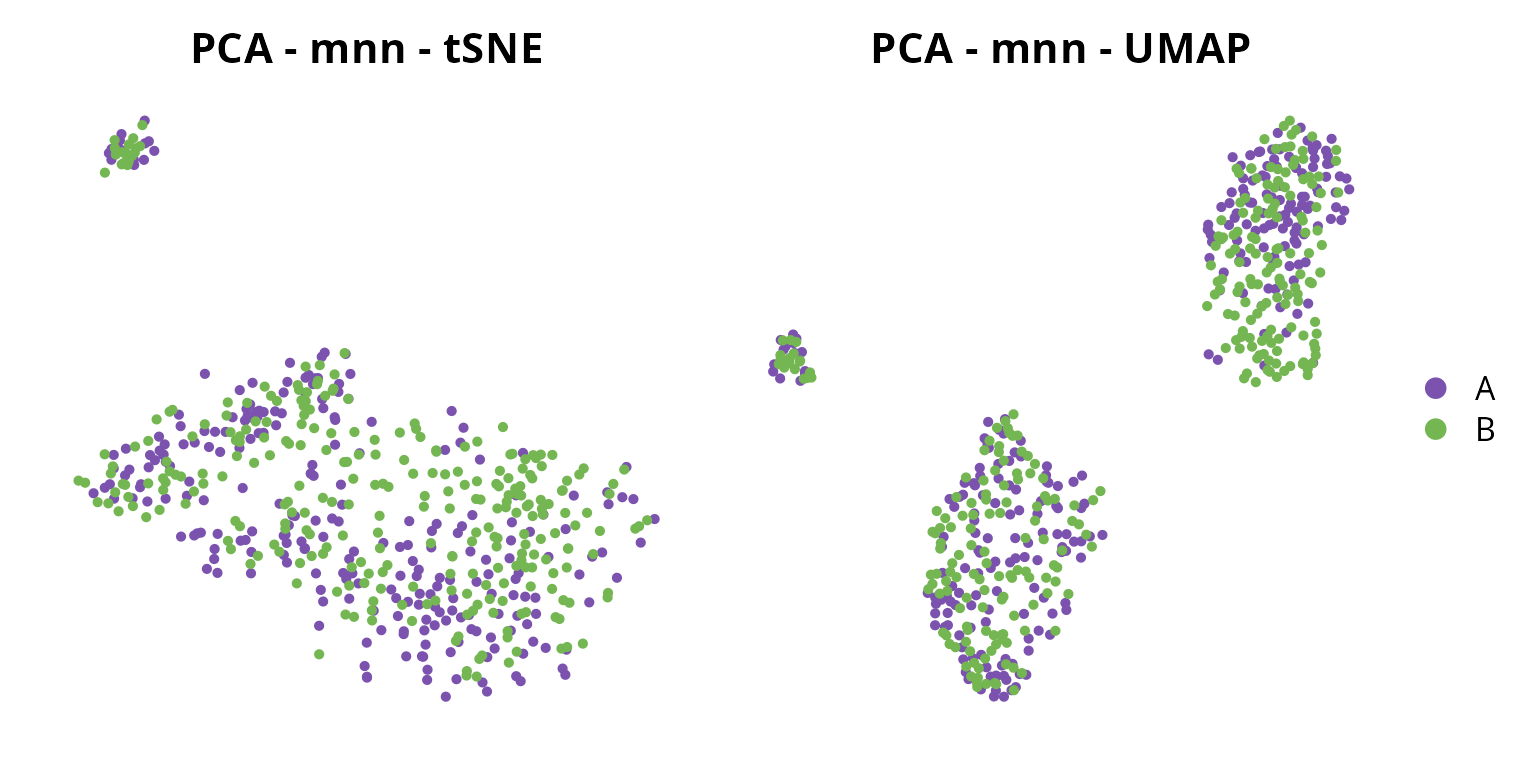
Diffusion map
We generate a diffusion map from the batch-effect corrected space:
reduction_name = "mnn"
sobj = aquarius::run_diffusion_map(sobj = sobj,
input = reduction_name,
n_eigs = 50)
sobj## An object of class Seurat
## 14361 features across 537 samples within 2 assays
## Active assay: RNA (12361 features, 2000 variable features)
## 3 layers present: scale.data, data, counts
## 1 other assay present: mnn.reconstructed
## 7 dimensional reductions calculated: RNA_pca, RNA_pca_51_tsne, RNA_pca_51_umap, mnn, mnn_51_umap, mnn_51_tsne, mnn_dmWe visualize it:
dm_name = paste0(reduction_name, "_dm")
Seurat::DimPlot(sobj, reduction = dm_name,
group.by = "orig.ident") +
ggplot2::scale_color_manual(values = sample_info$color,
breaks = sample_info$sample_identifiant) +
ggplot2::labs(title = dm_name) +
ggplot2::theme(aspect.ratio = 1,
plot.title = element_text(hjust = 0.5)) +
Seurat::NoAxes()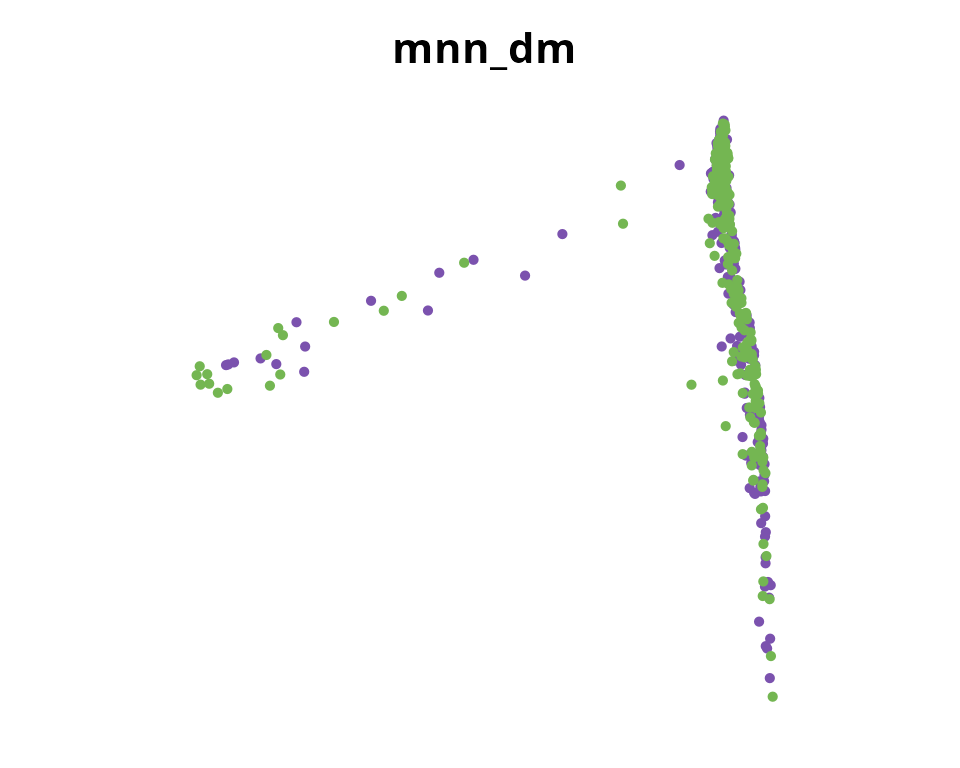
For visualisation, we keep the tSNE:
name2D = paste0("mnn_", ndims, "_tsne")Clustering
We generate a clustering:
sobj = Seurat::FindNeighbors(sobj, reduction = reduction_name, dims = 1:ndims)
sobj = Seurat::FindClusters(sobj, resolution = 0.5)## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 537
## Number of edges: 23208
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.7868
## Number of communities: 4
## Elapsed time: 0 seconds
clusters_plot = Seurat::DimPlot(sobj, reduction = name2D, label = TRUE) +
Seurat::NoAxes() + Seurat::NoLegend() +
ggplot2::labs(title = "Clusters ID") +
ggplot2::theme(aspect.ratio = 1,
plot.title = element_text(hjust = 0.5))
clusters_plot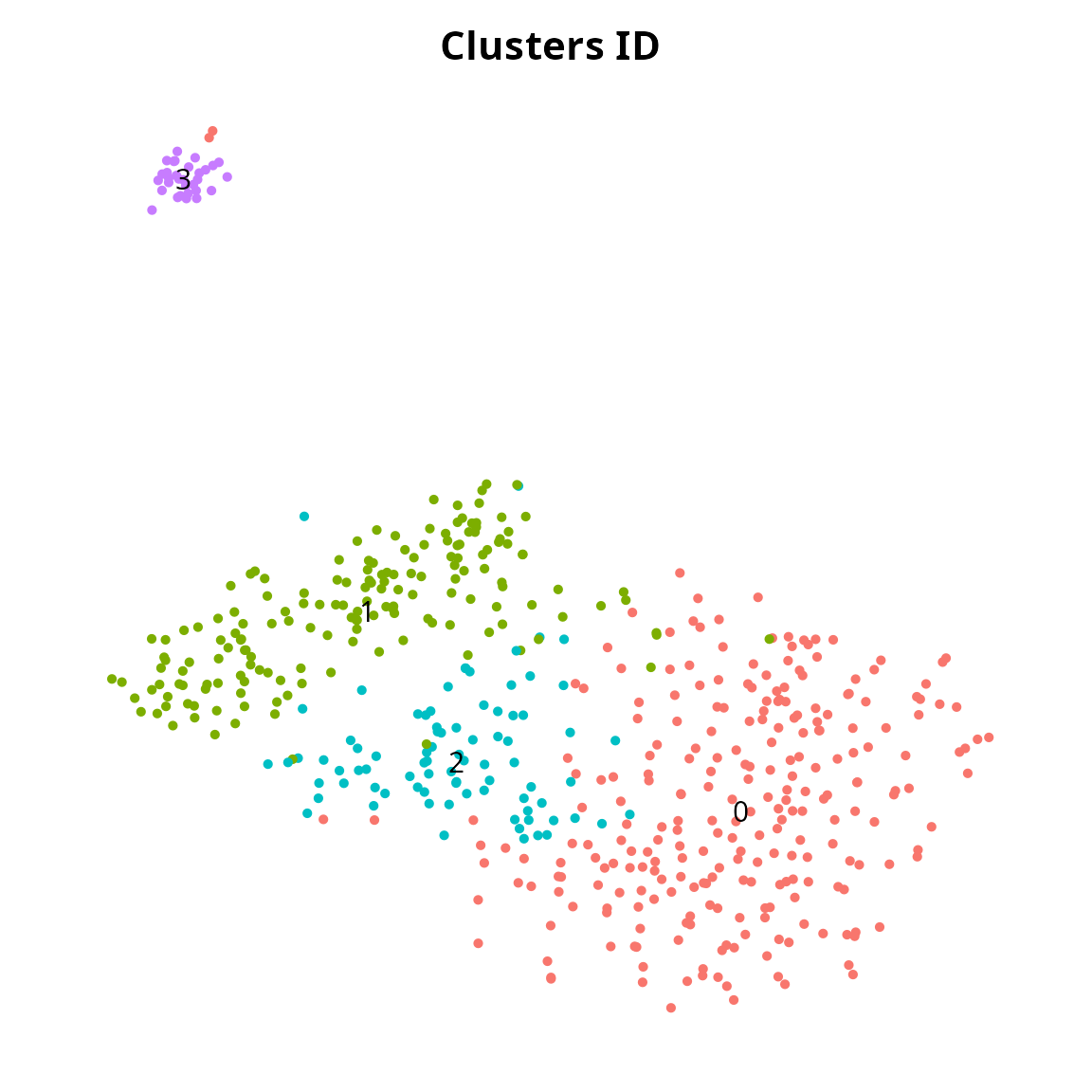
Visualization
We represent the 4 quality metrics:
plot_list = Seurat::FeaturePlot(sobj, reduction = name2D,
combine = FALSE, pt.size = 0.25,
features = c("percent.mt", "percent.rb", "log_nCount_RNA", "nFeature_RNA"))
plot_list = lapply(plot_list, FUN = function(one_plot) {
one_plot +
Seurat::NoAxes() +
ggplot2::scale_color_gradientn(colors = aquarius::palette_GrOrBl) +
ggplot2::theme(aspect.ratio = 1)
})
patchwork::wrap_plots(plot_list, nrow = 1)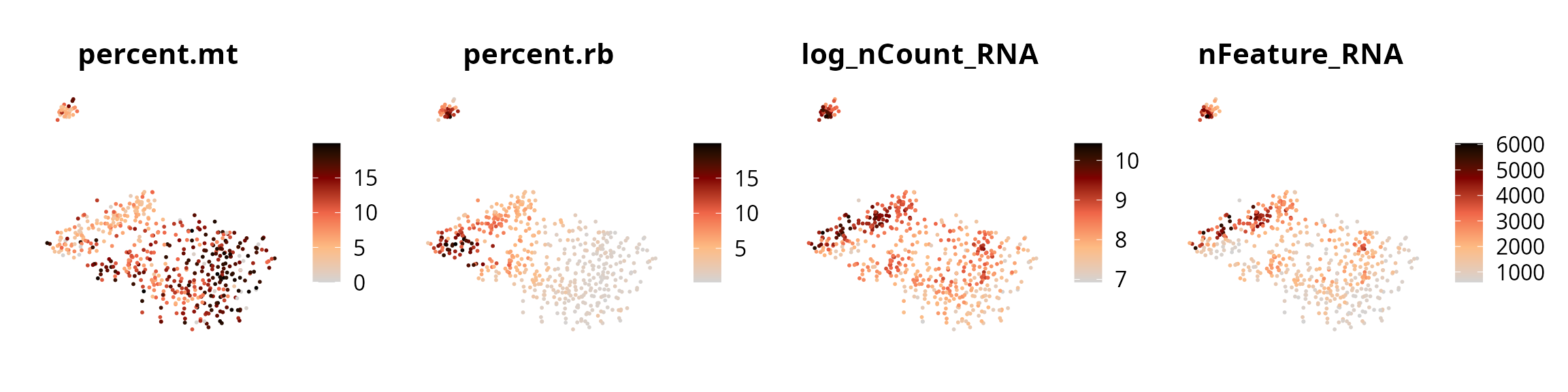
R Session
show
## R version 4.4.3 (2025-02-28)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.10
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.12.0
## LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=fr_FR.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=fr_FR.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=fr_FR.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=fr_FR.UTF-8 LC_IDENTIFICATION=C
##
## time zone: Europe/Paris
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] future_1.40.0 ggplot2_3.5.2 patchwork_1.3.0 dplyr_1.1.4
##
## loaded via a namespace (and not attached):
## [1] RcppAnnoy_0.0.22 splines_4.4.3
## [3] later_1.4.2 batchelor_1.22.0
## [5] tibble_3.2.1 polyclip_1.10-7
## [7] xts_0.14.1 fastDummies_1.7.5
## [9] lifecycle_1.0.4 doParallel_1.0.17
## [11] globals_0.17.0 lattice_0.22-6
## [13] MASS_7.3-65 vcd_1.4-13
## [15] magrittr_2.0.3 plotly_4.10.4
## [17] sass_0.4.10 rmarkdown_2.29
## [19] jquerylib_0.1.4 yaml_2.3.10
## [21] httpuv_1.6.16 Seurat_5.3.0
## [23] sctransform_0.4.2 spam_2.11-1
## [25] sp_2.2-0 spatstat.sparse_3.1-0
## [27] reticulate_1.42.0 cowplot_1.1.3
## [29] pbapply_1.7-2 RColorBrewer_1.1-3
## [31] ResidualMatrix_1.16.0 abind_1.4-8
## [33] zlibbioc_1.52.0 Rtsne_0.17
## [35] GenomicRanges_1.58.0 purrr_1.0.4
## [37] BiocGenerics_0.52.0 nnet_7.3-20
## [39] circlize_0.4.16 GenomeInfoDbData_1.2.13
## [41] IRanges_2.40.1 S4Vectors_0.44.0
## [43] ggrepel_0.9.6 irlba_2.3.5.1
## [45] listenv_0.9.1 spatstat.utils_3.1-3
## [47] goftest_1.2-3 RSpectra_0.16-2
## [49] spatstat.random_3.3-3 fitdistrplus_1.2-2
## [51] parallelly_1.43.0 pkgdown_2.1.2
## [53] DelayedMatrixStats_1.28.1 smoother_1.3
## [55] codetools_0.2-20 DelayedArray_0.32.0
## [57] scuttle_1.16.0 tidyselect_1.2.1
## [59] shape_1.4.6.1 UCSC.utils_1.2.0
## [61] farver_2.1.2 ScaledMatrix_1.14.0
## [63] matrixStats_1.5.0 stats4_4.4.3
## [65] spatstat.explore_3.4-2 jsonlite_2.0.0
## [67] GetoptLong_1.0.5 BiocNeighbors_2.0.1
## [69] e1071_1.7-16 Formula_1.2-5
## [71] progressr_0.15.1 ggridges_0.5.6
## [73] survival_3.8-3 iterators_1.0.14
## [75] systemfonts_1.2.3 foreach_1.5.2
## [77] tools_4.4.3 ragg_1.4.0
## [79] ica_1.0-3 Rcpp_1.0.14
## [81] glue_1.8.0 gridExtra_2.3
## [83] SparseArray_1.6.2 ranger_0.17.0
## [85] laeken_0.5.3 xfun_0.52
## [87] TTR_0.24.4 MatrixGenerics_1.18.1
## [89] ggthemes_5.1.0 GenomeInfoDb_1.42.3
## [91] withr_3.0.2 fastmap_1.2.0
## [93] boot_1.3-31 VIM_6.2.2
## [95] digest_0.6.37 rsvd_1.0.5
## [97] R6_2.6.1 mime_0.13
## [99] textshaping_1.0.1 colorspace_2.1-1
## [101] scattermore_1.2 tensor_1.5
## [103] spatstat.data_3.1-6 hexbin_1.28.5
## [105] tidyr_1.3.1 generics_0.1.3
## [107] data.table_1.17.0 robustbase_0.99-4-1
## [109] class_7.3-23 httr_1.4.7
## [111] htmlwidgets_1.6.4 S4Arrays_1.6.0
## [113] scatterplot3d_0.3-44 uwot_0.2.3
## [115] pkgconfig_2.0.3 gtable_0.3.6
## [117] ComplexHeatmap_2.23.1 lmtest_0.9-40
## [119] SingleCellExperiment_1.28.1 XVector_0.46.0
## [121] destiny_3.20.0 htmltools_0.5.8.1
## [123] carData_3.0-5 dotCall64_1.2
## [125] clue_0.3-66 SeuratObject_5.1.0
## [127] scales_1.4.0 Biobase_2.66.0
## [129] png_0.1-8 knn.covertree_1.0
## [131] spatstat.univar_3.1-2 knitr_1.50
## [133] reshape2_1.4.4 rjson_0.2.23
## [135] curl_6.2.2 aquarius_1.0.0
## [137] nlme_3.1-168 proxy_0.4-27
## [139] cachem_1.1.0 zoo_1.8-14
## [141] GlobalOptions_0.1.2 stringr_1.5.1
## [143] KernSmooth_2.23-26 parallel_4.4.3
## [145] miniUI_0.1.2 desc_1.4.3
## [147] pillar_1.10.2 grid_4.4.3
## [149] vctrs_0.6.5 pcaMethods_1.98.0
## [151] RANN_2.6.2 promises_1.3.2
## [153] car_3.1-3 BiocSingular_1.22.0
## [155] beachmat_2.22.0 xtable_1.8-4
## [157] cluster_2.1.8.1 evaluate_1.0.3
## [159] cli_3.6.5 compiler_4.4.3
## [161] rlang_1.1.6 crayon_1.5.3
## [163] future.apply_1.11.3 labeling_0.4.3
## [165] plyr_1.8.9 fs_1.6.6
## [167] stringi_1.8.7 viridisLite_0.4.2
## [169] deldir_2.0-4 BiocParallel_1.40.2
## [171] lazyeval_0.2.2 spatstat.geom_3.3-6
## [173] Matrix_1.7-3 RcppHNSW_0.6.0
## [175] RcppEigen_0.3.4.0.2 sparseMatrixStats_1.18.0
## [177] shiny_1.10.0 SummarizedExperiment_1.36.0
## [179] ROCR_1.0-11 igraph_2.1.4
## [181] bslib_0.9.0 DEoptimR_1.1-3-1
## [183] ggplot.multistats_1.0.1