Analysis
Trajectory inference
2025-05-03
Source:vignettes/examples/3_analysis/1_trajectory.Rmd
1_trajectory.RmdThis notebook shows the use of these functions from
aquarius:
aquarius::traj_inferenceaquarius::traj_tingaaquarius::plot_label_dimplotaquarius::plot_selectoraquarius::find_features_order
The goal of this script is to merge perform trajectory inference and use related functions.
## [1] "/usr/lib/R/library" "/usr/local/lib/R/site-library"
## [3] "/usr/lib/R/site-library"Preparation
In this section, we set the global settings of the analysis. We will store data there:
out_dir = "."The input dataset is there:
save_name = "tumor_cells"
data_path = paste0(out_dir, "/../2_combined/", save_name, "_sobj.rds")
data_path## [1] "./../2_combined/tumor_cells_sobj.rds"We define custom colors for each sample:
sample_info = data.frame(
project_name = c("A", "B"),
sample_identifier = c("A", "B"),
color = c("#7B52AE", "#74B652"),
row.names = c("A", "B"))
aquarius::plot_df(sample_info)
Load dataset
We load the dataset:
sobj = readRDS(data_path)
sobj## An object of class Seurat
## 14361 features across 537 samples within 2 assays
## Active assay: RNA (12361 features, 2000 variable features)
## 3 layers present: scale.data, data, counts
## 1 other assay present: mnn.reconstructed
## 7 dimensional reductions calculated: RNA_pca, RNA_pca_51_tsne, RNA_pca_51_umap, mnn, mnn_51_umap, mnn_51_tsne, mnn_dmWe set the projection of interest:
name2D = "mnn_dm"Visualization
We visualize the expression levels of some genes on the diffusion map:
x_lim = range(sobj@reductions[[name2D]]@cell.embeddings[, 1])
y_lim = range(sobj@reductions[[name2D]]@cell.embeddings[, 2])
features_oi = c("nFeature_RNA", "Mbp", "Mgp", "Birc5")
plot_list = lapply(features_oi, FUN = function(one_gene) {
p = Seurat::FeaturePlot(sobj, reduction = name2D,
pt.size = 0.25, order = TRUE,
features = one_gene) +
ggplot2::lims(x = x_lim, y = y_lim) +
Seurat::NoAxes() +
ggplot2::scale_color_gradientn(colors = aquarius::palette_GrOrBl) +
ggplot2::theme(aspect.ratio = 1)
return(p)
})
patchwork::wrap_plots(plot_list, nrow = 1)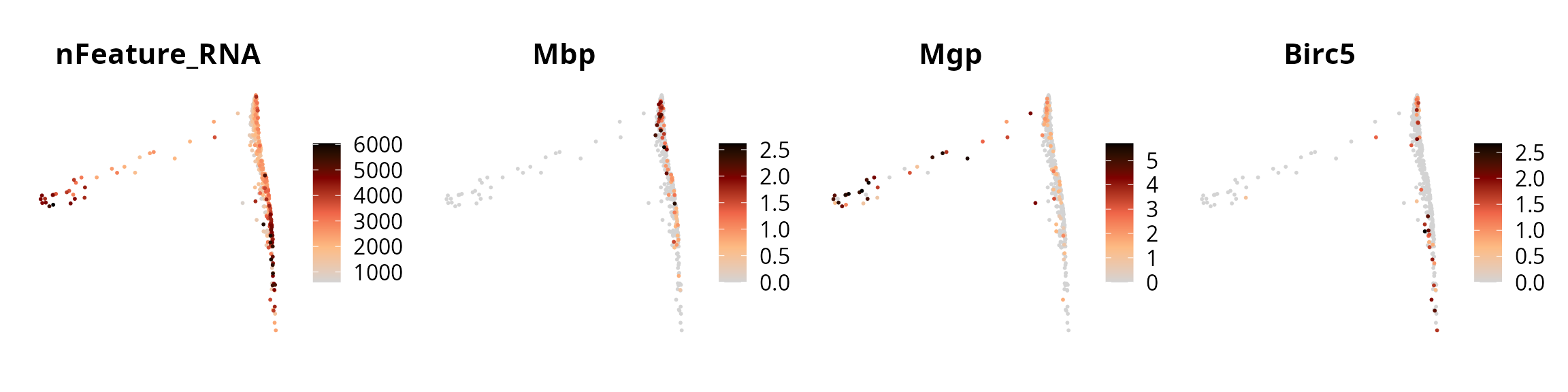
We visualize an interactive plot:
aquarius::plot_selector(sobj,
info_to_plot = "Mgp",
reduction = name2D)Trajectory inference
Using TInGa
We set the root manually:
root_cell_id = "B_\"CCATCACTCGTGGCTG-1\""
sobj$is_root = colnames(sobj) == root_cell_id
sobj$cell_name = colnames(sobj)
root_plot = aquarius::plot_label_dimplot(sobj,
reduction = name2D,
col_by = "is_root",
col_color = c("gray80", "red"),
label_by = "cell_name",
label_val = root_cell_id) +
ggplot2::labs(title = "Root cell") +
ggplot2::theme(plot.title = element_text(hjust = 0.5, face = "bold"),
aspect.ratio = 1) +
Seurat::NoLegend() + Seurat::NoAxes()
sobj$is_root = NULL
sobj$cell_name = NULL
root_plot
We perform trajectory inference:
traj_tinga = aquarius::traj_tinga(sobj,
expression_assay = "RNA",
expression_slot = "data",
count_assay = "RNA",
count_slot = "counts",
dimred_name = "mnn",
dimred_max_dim = 50,
root_cell_id = root_cell_id)
class(traj_tinga)## [1] "dynwrap::with_dimred" "dynwrap::with_trajectory"
## [3] "dynwrap::data_wrapper" "list"We add the pseudotime in the Seurat object:
sobj$pseudotime = traj_tinga$pseudotimeWe visualize the pseudotime using Seurat:
Seurat::FeaturePlot(sobj, features = "pseudotime",
reduction = name2D) +
ggplot2::scale_color_gradientn(colors = viridis::viridis(n = 100)) +
ggplot2::lims(x = x_lim, y = y_lim) +
ggplot2::labs(title = "Pseudotime") +
ggplot2::theme(aspect.ratio = 1,
plot.title = element_text(hjust = 0.5)) +
Seurat::NoAxes()
We visualize the pseudotime using dynplot:
dynplot::plot_dimred(trajectory = traj_tinga,
label_milestones = FALSE,
plot_trajectory = TRUE,
dimred = sobj[[name2D]]@cell.embeddings,
color_cells = 'pseudotime',
color_trajectory = "none")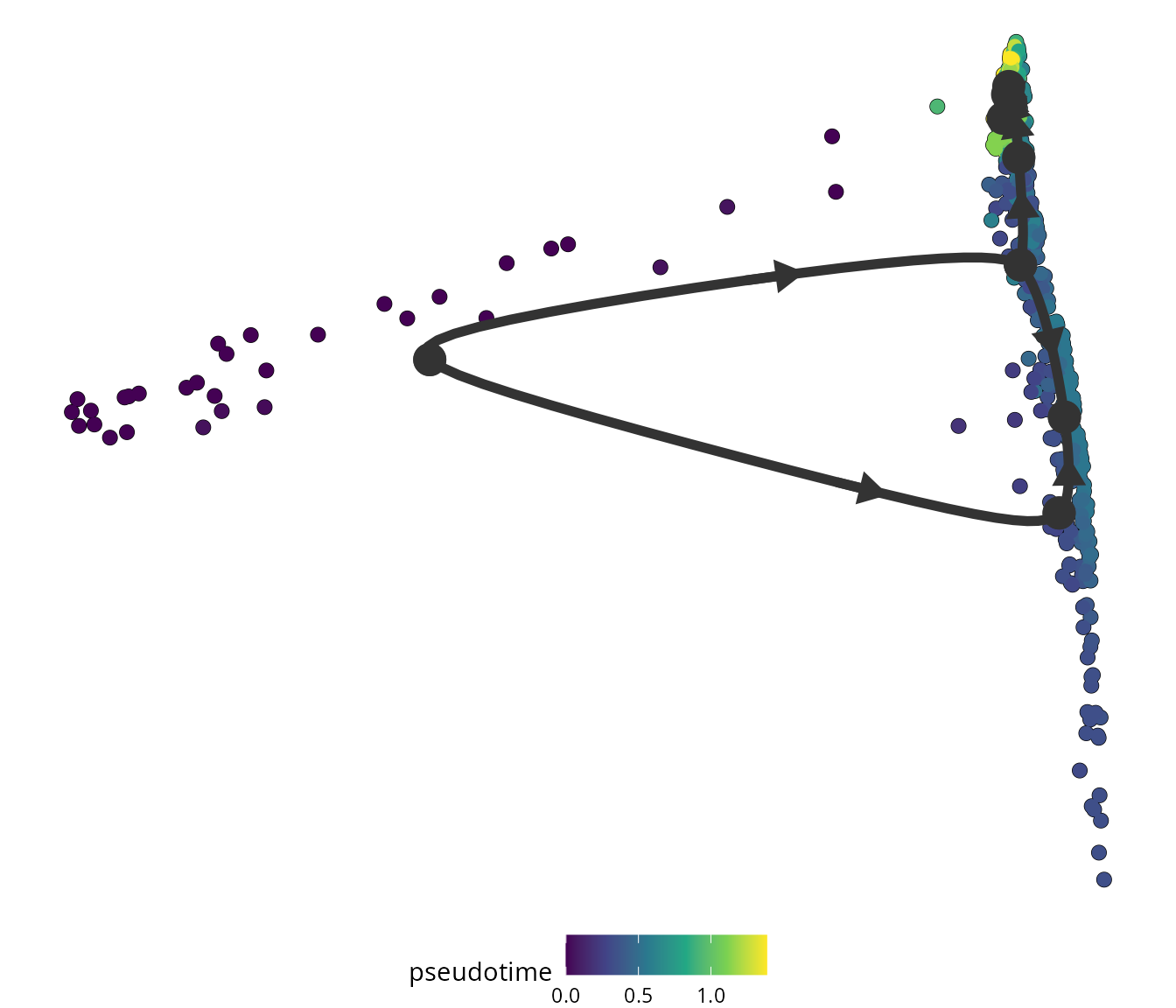
Using slingshot
We perform trajectory inference:
input_data = sobj@reductions[[name2D]]@cell.embeddings[, c(1:50)]
traj_slingshot = slingshot::slingshot(data = input_data,
clusterLabels = sobj$seurat_clusters,
start.clus = sobj$seurat_clusters[root_cell_id])
class(traj_slingshot)## [1] "PseudotimeOrdering"
## attr(,"package")
## [1] "TrajectoryUtils"Here is another way to do the same (eval = FALSE). But,
it is time consuming as it uses a software container.
traj_slingshot = aquarius::traj_inference(sobj,
expression_assay = "RNA",
expression_slot = "data",
count_assay = "RNA",
count_slot = "counts",
dimred_name = name2D,
dimred_max_dim = 50,
root_cell_id = root_cell_id,
ti_method = dynmethods::ti_slingshot())
class(traj_slingshot)How many trajectories are available ?
head(slingshot::slingPseudotime(traj_slingshot))## Lineage1
## A_"AAACCCATCAAGTCGT-1" 0.5742917
## A_"AACACACCAGTAACAA-1" 0.6774816
## A_"AACGAAACAGTCACGC-1" 0.6615958
## A_"AACTTCTAGGGCCCTT-1" 0.6344614
## A_"AAGCGAGGTATCCCAA-1" 0.5974519
## A_"AAGTCGTAGTCACTAC-1" 0.6384976We add the associated pseudotime in the Seurat object:
sobj$pseudotime = slingshot::slingPseudotime(traj_slingshot)[, 1][colnames(sobj)]We visualize the pseudotime using Seurat:
Seurat::FeaturePlot(sobj, features = "pseudotime",
reduction = name2D) +
ggplot2::scale_color_gradientn(colors = viridis::viridis(n = 100)) +
ggplot2::lims(x = x_lim, y = y_lim) +
ggplot2::labs(title = "Pseudotime") +
ggplot2::theme(aspect.ratio = 1,
plot.title = element_text(hjust = 0.5)) +
Seurat::NoAxes()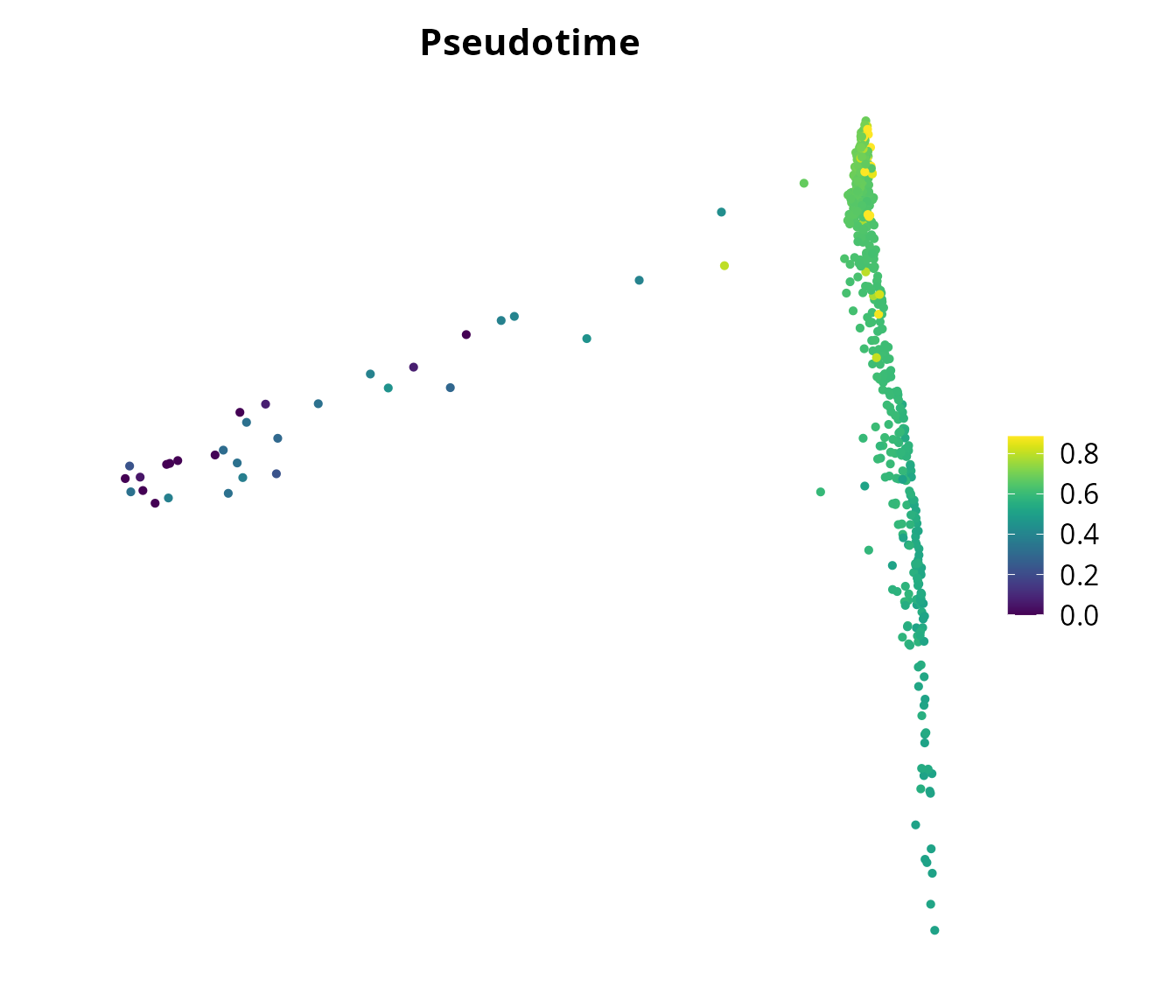
We visualize the pseudotime using the suggested method in
slingshot vignette.
# Reduction
rd = sobj[[name2D]]@cell.embeddings[, c(1:2)]
# Pseudotime
pdt = sobj$pseudotime
# Colors
colors = viridis::viridis(n = 100) # discrete
colors_plot = colors[cut(pdt, breaks = 100)]
# Plot
plot(rd,
col = colors_plot,
pch = 16,
asp = 1)
lines(slingshot::SlingshotDataSet(traj_slingshot),
lwd = 2,
col = 'black')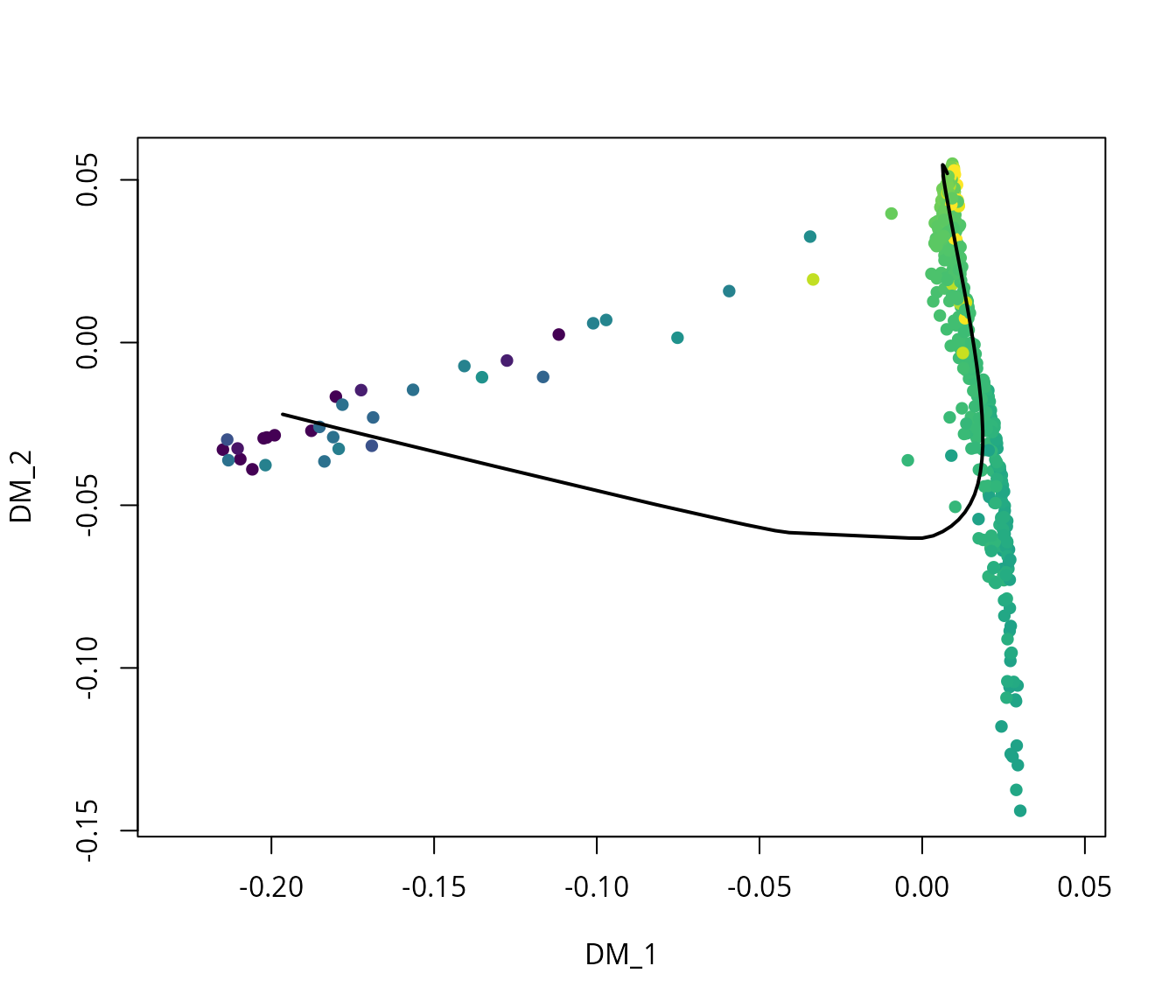
Heatmap
We identified genes of interest:
features = c("Mbp", "Dcn", "Birc5", "S100b", "Mgp")We define a matrix to make a heatmap from:
mat_expression = Seurat::GetAssayData(sobj, assay = "RNA", slot = "data")
mat_expression = mat_expression[features, ]
mat_expression = Matrix::t(mat_expression)
mat_expression = dynutils::scale_quantile(mat_expression) # between 0 and 1
mat_expression = Matrix::t(mat_expression)
mat_expression = Matrix::as.matrix(mat_expression)
dim(mat_expression)## [1] 5 537We define custom colors:
list_colors = list()
list_colors[["expression"]] = aquarius::palette_BlWhRd
list_colors[["pseudotime"]] = circlize::colorRamp2(breaks = seq(min(sobj$pseudotime),
max(sobj$pseudotime),
length.out = 9),
colors = viridis::viridis(9))We prepare the heatmap annotation:
ha_top = ComplexHeatmap::HeatmapAnnotation(
pseudotime = sobj$pseudotime,
annotation_legend_param = list(pseudotime = list(at = range(sobj$pseudotime),
labels = c("min", "max"))),
col = list(pseudotime = list_colors[["pseudotime"]]))Automatic order
We build a heatmap:
ht = ComplexHeatmap::Heatmap(
mat_expression,
# Expression colors
col = list_colors[["expression"]],
heatmap_legend_param = list(title = "expression", at = c(0, 1),
labels = c("low", "high")),
# Annotation
top_annotation = ha_top,
# Cells order
column_order = order(sobj$pseudotime),
cluster_columns = FALSE,
# Cells label
show_column_names = FALSE,
# Gene grouping
cluster_rows = TRUE,
show_row_dend = TRUE,
# Gene labels
row_labels = features,
show_row_names = TRUE,
row_names_gp = grid::gpar(fontsize = 12),
# Visual aspect
show_heatmap_legend = TRUE,
border = TRUE,
use_raster = FALSE)We draw it:
ComplexHeatmap::draw(ht, # legend_grouping = "original",
merge_legend = TRUE,
heatmap_legend_side = "bottom",
annotation_legend_side = "bottom")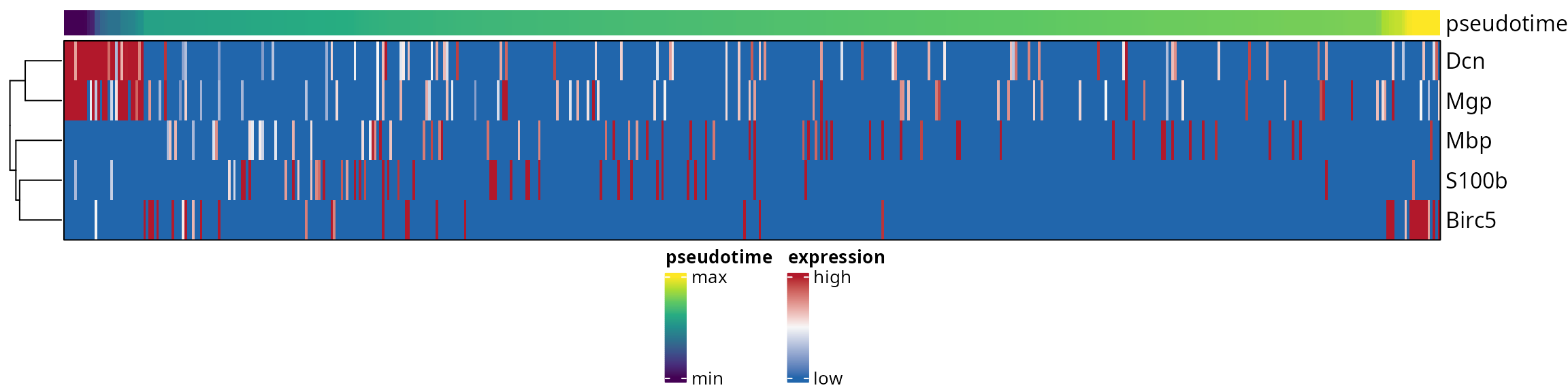
Pseudotime-consistent order
We identify an order to represent genes along pseudotime. First, we define a suited dataframe, containing the pseudotime and the genes of interest:
df_features = mat_expression %>%
t() %>%
as.data.frame.matrix()
df_features$pseudotime = sobj$pseudotime
head(df_features)## Mbp Dcn Birc5 S100b Mgp pseudotime
## A_"AAACCCATCAAGTCGT-1" 0.0000000 0 0 1 0 0.5742917
## A_"AACACACCAGTAACAA-1" 0.0000000 0 0 0 0 0.6774816
## A_"AACGAAACAGTCACGC-1" 0.0000000 0 0 0 0 0.6615958
## A_"AACTTCTAGGGCCCTT-1" 0.0000000 0 0 0 0 0.6344614
## A_"AAGCGAGGTATCCCAA-1" 0.7775097 0 0 1 0 0.5974519
## A_"AAGTCGTAGTCACTAC-1" 1.0000000 0 0 0 0 0.6384976We use the aquarius::find_features_order function to
identify a beautiful order among genes:
features_order = aquarius::find_features_order(df_features)
features_order## [1] "Mgp" "Dcn" "S100b" "Mbp" "Birc5"Now we can make the heatmap by forcing this ordering:
ht = ComplexHeatmap::Heatmap(
mat_expression[features_order, ], # <---- changed
# Expression colors
col = list_colors[["expression"]],
heatmap_legend_param = list(title = "expression", at = c(0, 1),
labels = c("low", "high")),
# Annotation
top_annotation = ha_top,
# Cells order
column_order = order(sobj$pseudotime),
cluster_columns = FALSE,
# Cells label
show_column_names = FALSE,
# Gene grouping
cluster_rows = FALSE, # <---- changed
show_row_dend = FALSE, # <---- changed
# Gene labels
row_labels = features_order, # <---- changed
show_row_names = TRUE,
row_names_gp = grid::gpar(fontsize = 12),
# Visual aspect
show_heatmap_legend = TRUE,
border = TRUE,
use_raster = FALSE)We draw it:
ComplexHeatmap::draw(ht, # legend_grouping = "original",
merge_legend = TRUE,
heatmap_legend_side = "bottom",
annotation_legend_side = "bottom")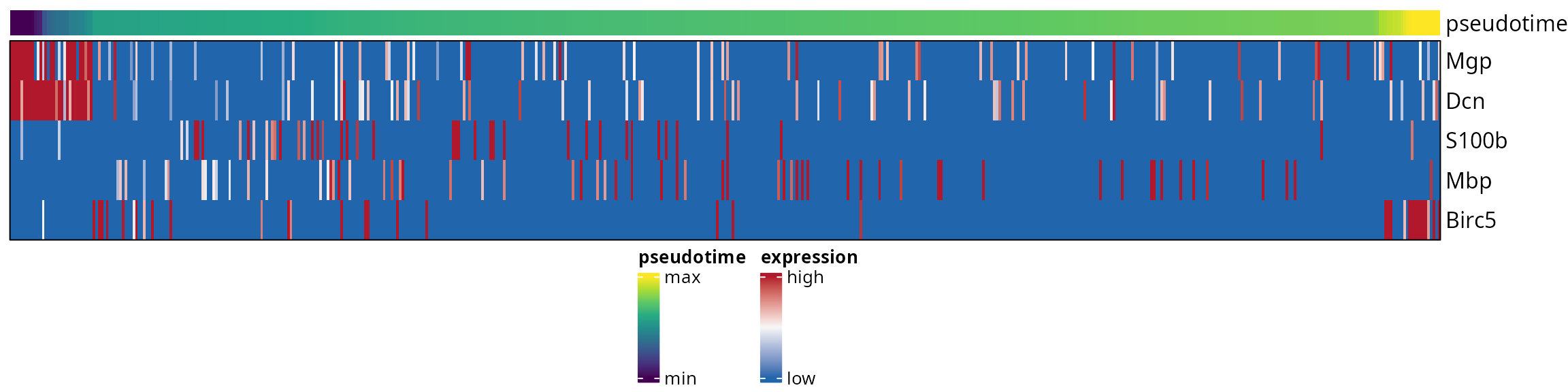
R Session
show
## R version 4.4.3 (2025-02-28)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.10
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.12.0
## LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=fr_FR.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=fr_FR.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=fr_FR.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=fr_FR.UTF-8 LC_IDENTIFICATION=C
##
## time zone: Europe/Paris
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] dynutils_1.0.11 dynwrap_1.2.4 purrr_1.0.4 tidyr_1.3.1
## [5] ggplot2_3.5.2 patchwork_1.3.0 dplyr_1.1.4
##
## loaded via a namespace (and not attached):
## [1] fs_1.6.6 matrixStats_1.5.0
## [3] spatstat.sparse_3.1-0 httr_1.4.7
## [5] RColorBrewer_1.1-3 doParallel_1.0.17
## [7] tools_4.4.3 sctransform_0.4.2
## [9] R6_2.6.1 lazyeval_0.2.2
## [11] uwot_0.2.3 GetoptLong_1.0.5
## [13] withr_3.0.2 sp_2.2-0
## [15] gridExtra_2.3 progressr_0.15.1
## [17] cli_3.6.5 Biobase_2.66.0
## [19] textshaping_1.0.1 Cairo_1.6-2
## [21] aquarius_1.0.0 spatstat.explore_3.4-2
## [23] fastDummies_1.7.5 labeling_0.4.3
## [25] sass_0.4.10 Seurat_5.3.0
## [27] spatstat.data_3.1-6 readr_2.1.5
## [29] ggridges_0.5.6 pbapply_1.7-2
## [31] pkgdown_2.1.2 slingshot_2.14.0
## [33] systemfonts_1.2.3 parallelly_1.43.0
## [35] GA_3.2.4 TInGa_0.0.0.9000
## [37] visNetwork_2.1.2 generics_0.1.3
## [39] shape_1.4.6.1 ica_1.0-3
## [41] spatstat.random_3.3-3 crosstalk_1.2.1
## [43] Matrix_1.7-3 S4Vectors_0.44.0
## [45] abind_1.4-8 lifecycle_1.0.4
## [47] yaml_2.3.10 SummarizedExperiment_1.36.0
## [49] SparseArray_1.6.2 Rtsne_0.17
## [51] grid_4.4.3 promises_1.3.2
## [53] crayon_1.5.3 miniUI_0.1.2
## [55] lattice_0.22-6 cowplot_1.1.3
## [57] dynfeature_1.0.1 pillar_1.10.2
## [59] knitr_1.50 ComplexHeatmap_2.23.1
## [61] GenomicRanges_1.58.0 rjson_0.2.23
## [63] future.apply_1.11.3 codetools_0.2-20
## [65] glue_1.8.0 spatstat.univar_3.1-2
## [67] data.table_1.17.0 remotes_2.5.0
## [69] dynparam_1.0.2 vctrs_0.6.5
## [71] png_0.1-8 spam_2.11-1
## [73] testthat_3.2.3 gtable_0.3.6
## [75] assertthat_0.2.1 cachem_1.1.0
## [77] xfun_0.52 princurve_2.1.6
## [79] S4Arrays_1.6.0 mime_0.13
## [81] tidygraph_1.3.1 survival_3.8-3
## [83] randomForestSRC_3.3.3 SingleCellExperiment_1.28.1
## [85] iterators_1.0.14 DiagrammeR_1.0.11
## [87] fitdistrplus_1.2-2 ROCR_1.0-11
## [89] nlme_3.1-168 gng_0.1.0
## [91] RcppAnnoy_0.0.22 GenomeInfoDb_1.42.3
## [93] data.tree_1.1.0 bslib_0.9.0
## [95] dyneval_0.9.9 irlba_2.3.5.1
## [97] KernSmooth_2.23-26 colorspace_2.1-1
## [99] BiocGenerics_0.52.0 carrier_0.1.1
## [101] tidyselect_1.2.1 processx_3.8.6
## [103] proxyC_0.5.2 compiler_4.4.3
## [105] desc_1.4.3 DelayedArray_0.32.0
## [107] plotly_4.10.4 scales_1.4.0
## [109] lmtest_0.9-40 stringr_1.5.1
## [111] digest_0.6.37 goftest_1.2-3
## [113] spatstat.utils_3.1-3 rmarkdown_2.29
## [115] XVector_0.46.0 htmltools_0.5.8.1
## [117] pkgconfig_2.0.3 sparseMatrixStats_1.18.0
## [119] MatrixGenerics_1.18.1 fastmap_1.2.0
## [121] rlang_1.1.6 GlobalOptions_0.1.2
## [123] htmlwidgets_1.6.4 UCSC.utils_1.2.0
## [125] DelayedMatrixStats_1.28.1 shiny_1.10.0
## [127] farver_2.1.2 jquerylib_0.1.4
## [129] zoo_1.8-14 jsonlite_2.0.0
## [131] BiocParallel_1.40.2 magrittr_2.0.3
## [133] GenomeInfoDbData_1.2.13 dotCall64_1.2
## [135] dyndimred_1.0.4 Rcpp_1.0.14
## [137] viridis_0.6.5 reticulate_1.42.0
## [139] TrajectoryUtils_1.14.0 stringi_1.8.7
## [141] ggraph_2.2.1 brio_1.1.5
## [143] zlibbioc_1.52.0 MASS_7.3-65
## [145] dynplot_1.1.2 plyr_1.8.9
## [147] parallel_4.4.3 listenv_0.9.1
## [149] ggrepel_0.9.6 deldir_2.0-4
## [151] graphlayouts_1.2.2 splines_4.4.3
## [153] tensor_1.5 hms_1.1.3
## [155] circlize_0.4.16 ps_1.9.1
## [157] igraph_2.1.4 ranger_0.17.0
## [159] spatstat.geom_3.3-6 babelwhale_1.2.0
## [161] RcppHNSW_0.6.0 reshape2_1.4.4
## [163] stats4_4.4.3 evaluate_1.0.3
## [165] SeuratObject_5.1.0 tzdb_0.5.0
## [167] foreach_1.5.2 tweenr_2.0.3
## [169] httpuv_1.6.16 RANN_2.6.2
## [171] polyclip_1.10-7 future_1.40.0
## [173] clue_0.3-66 scattermore_1.2
## [175] lmds_0.1.0 ggforce_0.4.2
## [177] xtable_1.8-4 RSpectra_0.16-2
## [179] later_1.4.2 viridisLite_0.4.2
## [181] ragg_1.4.0 tibble_3.2.1
## [183] memoise_2.0.1 IRanges_2.40.1
## [185] cluster_2.1.8.1 globals_0.17.0