The goal of this script is to make figure to show how to use the
functions from aquarius.
## [1] "/usr/lib/R/library" "/usr/local/lib/R/site-library"
## [3] "/usr/lib/R/site-library"Preparation
We load the dataset:
sobj = readRDS("./2_combined/combined_sobj.rds")
sobj## An object of class Seurat
## 13774 features across 1108 samples within 1 assay
## Active assay: RNA (13774 features, 2000 variable features)
## 3 layers present: scale.data, data, counts
## 6 dimensional reductions calculated: RNA_pca, RNA_pca_48_tsne, RNA_pca_48_umap, harmony, harmony_48_umap, harmony_48_tsneHere are custom colors for each cell type:
color_markers = c("macrophages" = "#6ECEDF",
"tumor cells" = "#DA2328")We define custom colors for each sample:
sample_info = data.frame(
project_name = c("A", "B"),
sample_identifier = c("A", "B"),
color = c("#7B52AE", "#74B652"),
row.names = c("A", "B"))
aquarius::plot_df(sample_info)
Visualization
Each section is named by the function it refers to.
plot_alluvial
We want to visualize the changes between a single-cell level cell type annotation and the annotation smoothed at a cluster level. Before making the figure, we smooth the annotation.
sobj$cell_type = factor(sobj$cell_type, levels = names(color_markers))
cluster_type = table(sobj$cell_type, sobj$seurat_clusters) %>%
prop.table(., margin = 2) %>%
apply(., 2, which.max)
cluster_type = setNames(nm = names(cluster_type),
levels(sobj$cell_type)[cluster_type])
sobj$cluster_type = setNames(nm = colnames(sobj),
cluster_type[sobj$seurat_clusters])
sobj$cluster_type = factor(sobj$cluster_type, levels = names(color_markers))The alluvial plot shows how the annotation of cells changes between the two methods.
aquarius::plot_alluvial(sobj@meta.data,
column1 = "cell_type",
column2 = "cluster_type",
colors = color_markers)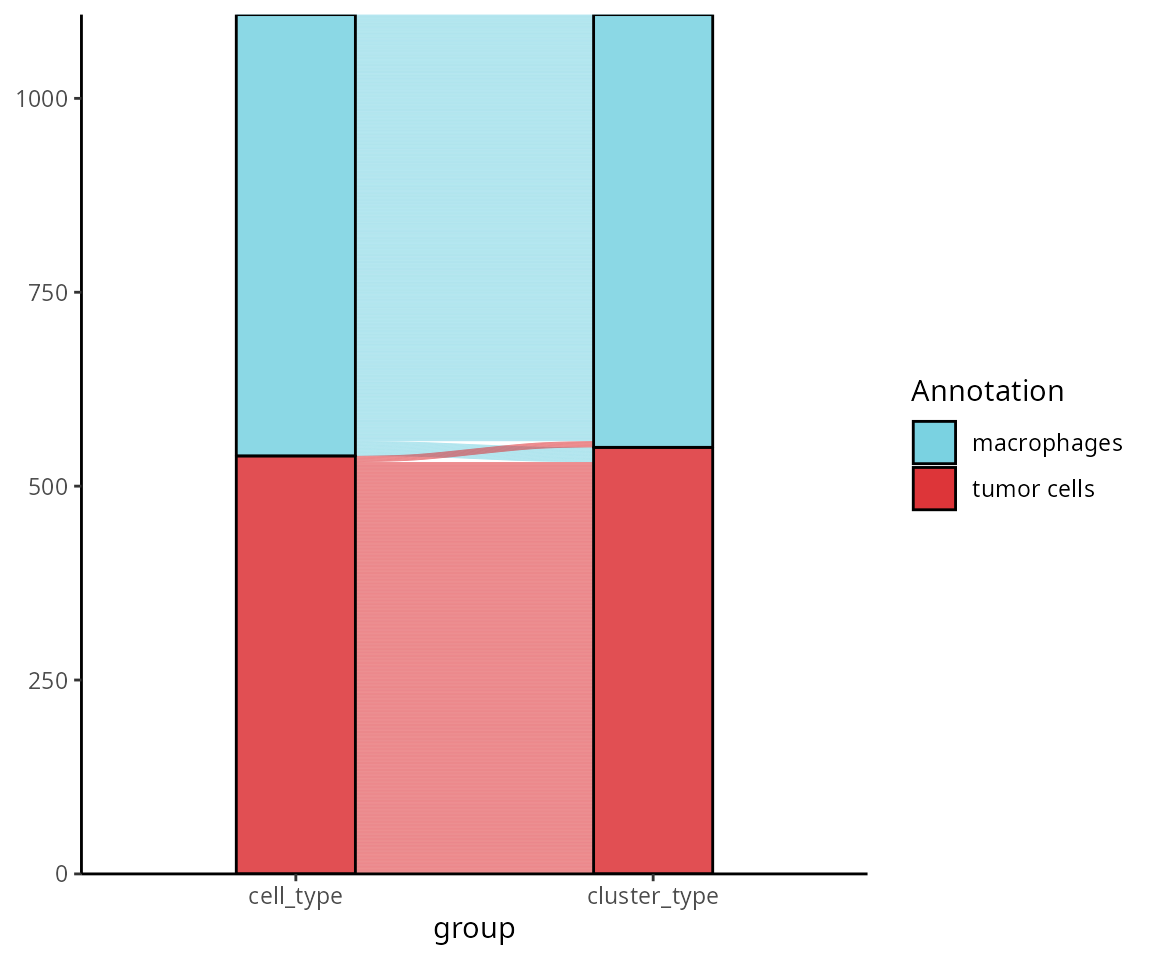
plot_c30_palette
We visualize the aquarius::c30_palette color
palette.
aquarius::plot_c30_palette()
The colors are:
aquarius::c30_palette## [1] "dodgerblue2" "#E31A1C" "green4" "#6A3D9A"
## [5] "#FF7F00" "black" "gold1" "skyblue2"
## [9] "#FB9A99" "palegreen2" "#CAB2D6" "#FDBF6F"
## [13] "gray85" "khaki2" "burlywood3" "orchid1"
## [17] "maroon1" "blue1" "steelblue4" "darkturquoise"
## [21] "green1" "yellow4" "yellow3" "darkorange4"
## [25] "brown" "darkmagenta" "plum3" "mediumseagreen"
## [29] "chocolate3" "gray55"plot_piechart_subpopulation
aquarius::plot_piechart_subpopulation(
sobj,
reduction = "harmony_48_tsne",
big_group_column = "cell_type",
big_group_of_interest = "macrophages",
big_group_color = color_markers[["macrophages"]],
small_group_column = "seurat_clusters",
composition_column = "orig.ident",
composition_color = sample_info$color,
bg_color = "gray92")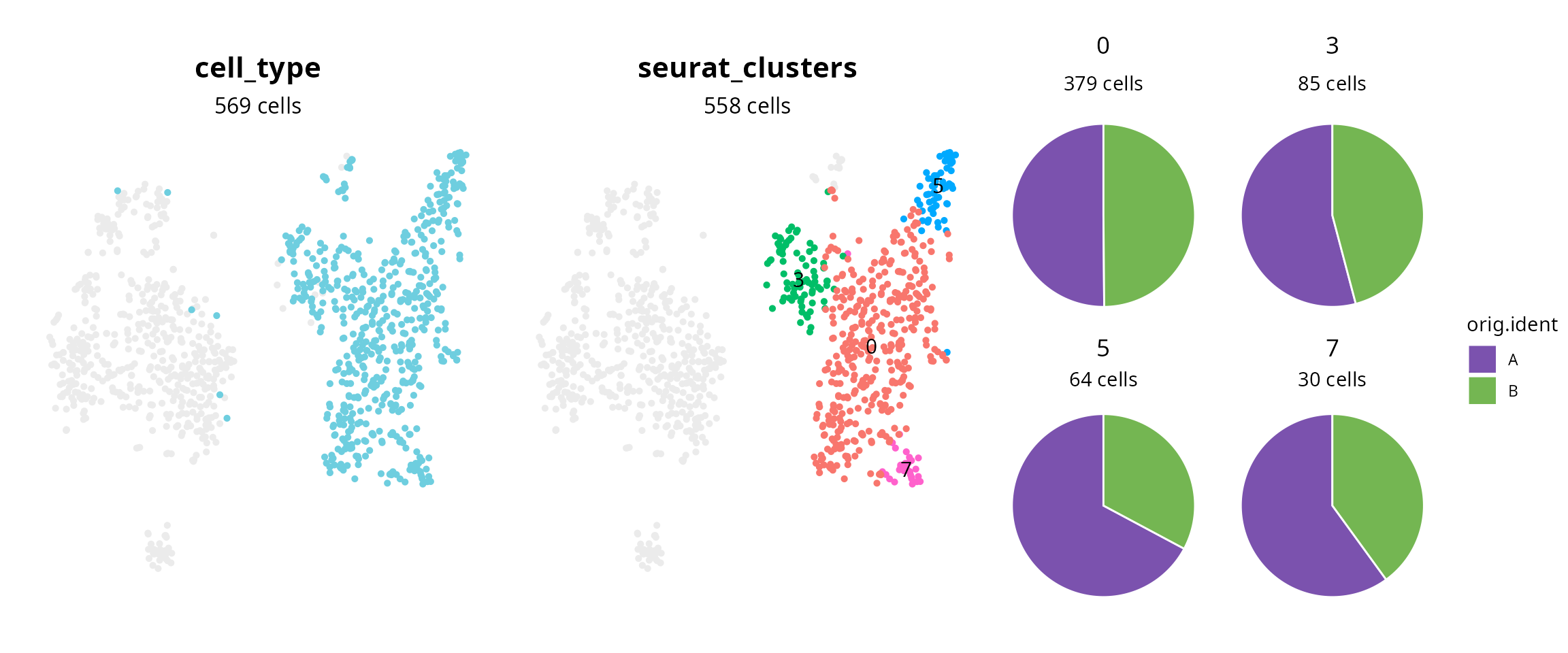
plot_subpopulation
Seurat::Idents(sobj) = sobj$cell_type
aquarius::plot_subpopulation(sobj,
reduction = "harmony_48_tsne",
identity = "tumor cells",
group_color = color_markers,
name_other = "other cells")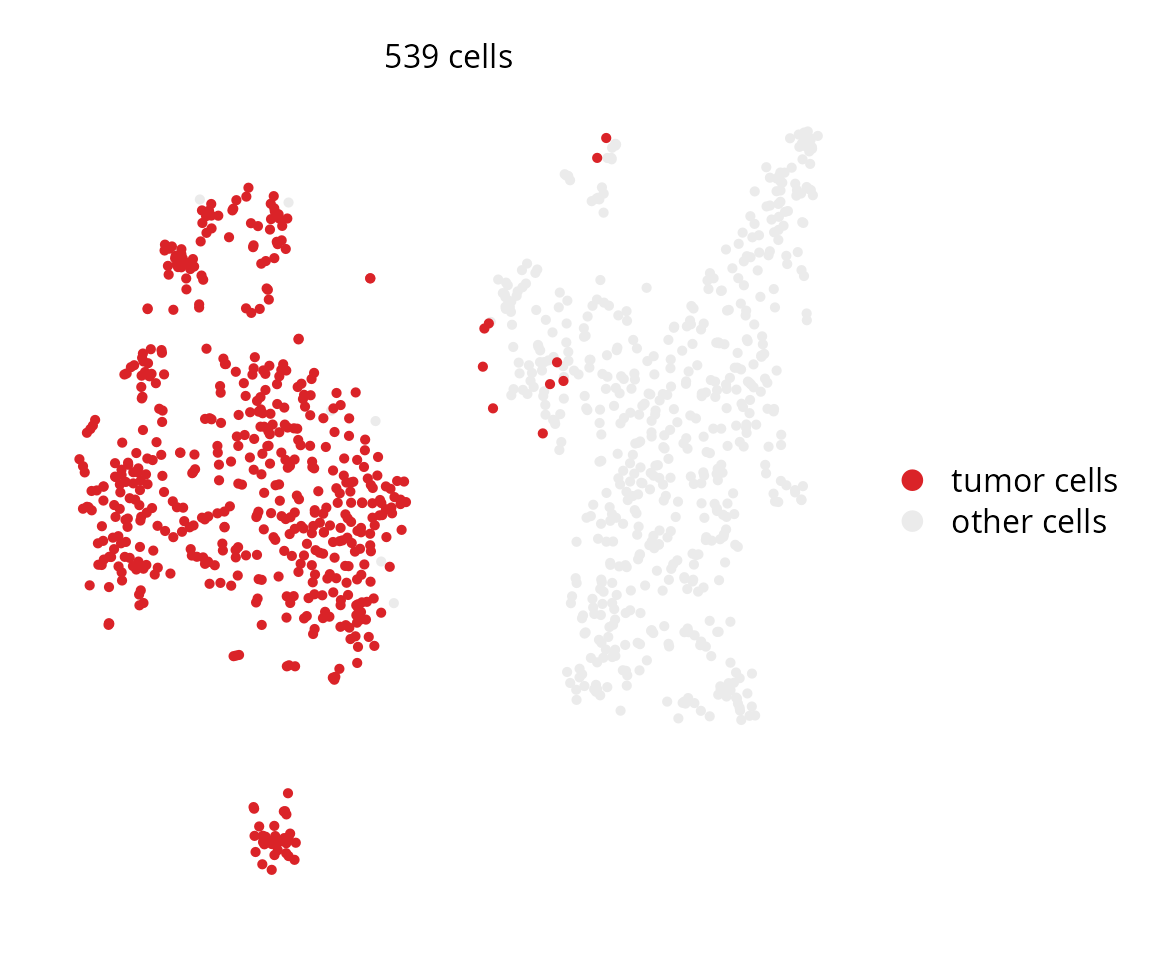
Other functions
Each section is named by the function it refers to.
print_to_copy_paste
A function to easily copy-paste a character vector:
my_vector = aquarius::c30_palette
aquarius::print_to_copy_paste(my_vector)## dodgerblue2
## #E31A1C
## green4
## #6A3D9A
## #FF7F00
## black
## gold1
## skyblue2
## #FB9A99
## palegreen2
## #CAB2D6
## #FDBF6F
## gray85
## khaki2
## burlywood3
## orchid1
## maroon1
## blue1
## steelblue4
## darkturquoise
## green1
## yellow4
## yellow3
## darkorange4
## brown
## darkmagenta
## plum3
## mediumseagreen
## chocolate3
## gray55run_rescale
We consider a numerical vector:
df = data.frame(original = sobj$score_macrophages)
head(df)## original
## A_"AAACCCATCAAGTCGT-1" -1.13779791
## A_"AAAGGTATCGTCTCAC-1" 0.96846694
## A_"AAAGTGACATCATTTC-1" 0.97000123
## A_"AAAGTGATCATGGAGG-1" -0.04234935
## A_"AACAAAGTCATTGTTC-1" 1.00857441
## A_"AACAAGAGTTTGATCG-1" 0.61423985We re-scale the distribution between -5 and 2, without keeping the 0 value at 0:
df$keep_zero_FALSE = aquarius::run_rescale(df$original,
new_min = -5,
new_max = 2,
keep_zero = FALSE)
ggplot2::ggplot(df, aes(x = original, y = keep_zero_FALSE)) +
ggplot2::geom_abline(slope = 1, intercept = 0, lty = 2, color = "gray") +
ggplot2::geom_hline(yintercept = 0, color = "gray") +
ggplot2::geom_vline(xintercept = 0, color = "gray") +
ggplot2::labs(x = "Original values",
y = "Rescaled values",
title = "aquarius::run_rescale",
subtitle = "with keep_zero = FALSE") +
ggplot2::geom_point() +
ggplot2::theme_classic() +
ggplot2::theme(plot.title = element_text(hjust = 0.5),
plot.subtitle = element_text(hjust = 0.5),
panel.grid.major = element_line())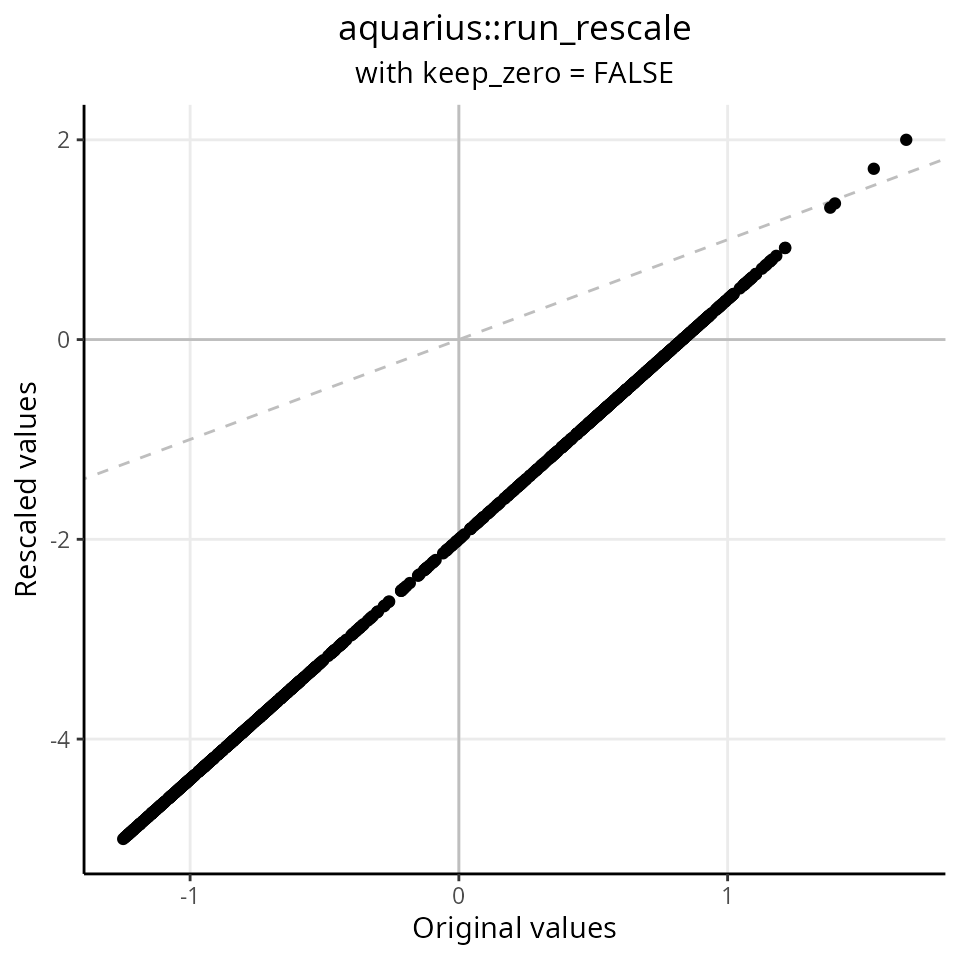
We re-scale the distribution between -5 and 2, with keeping the 0 value at 0:
df$keep_zero_TRUE = aquarius::run_rescale(df$original,
new_min = -5,
new_max = 2,
keep_zero = TRUE)
ggplot2::ggplot(df, aes(x = original, y = keep_zero_TRUE)) +
ggplot2::geom_abline(slope = 1, intercept = 0, lty = 2, color = "gray") +
ggplot2::geom_hline(yintercept = 0, color = "gray") +
ggplot2::geom_vline(xintercept = 0, color = "gray") +
ggplot2::labs(x = "Original values",
y = "Rescaled values",
title = "aquarius::run_rescale",
subtitle = "with keep_zero = TRUE") +
ggplot2::geom_point() +
ggplot2::theme_classic() +
ggplot2::theme(plot.title = element_text(hjust = 0.5),
plot.subtitle = element_text(hjust = 0.5),
panel.grid.major = element_line())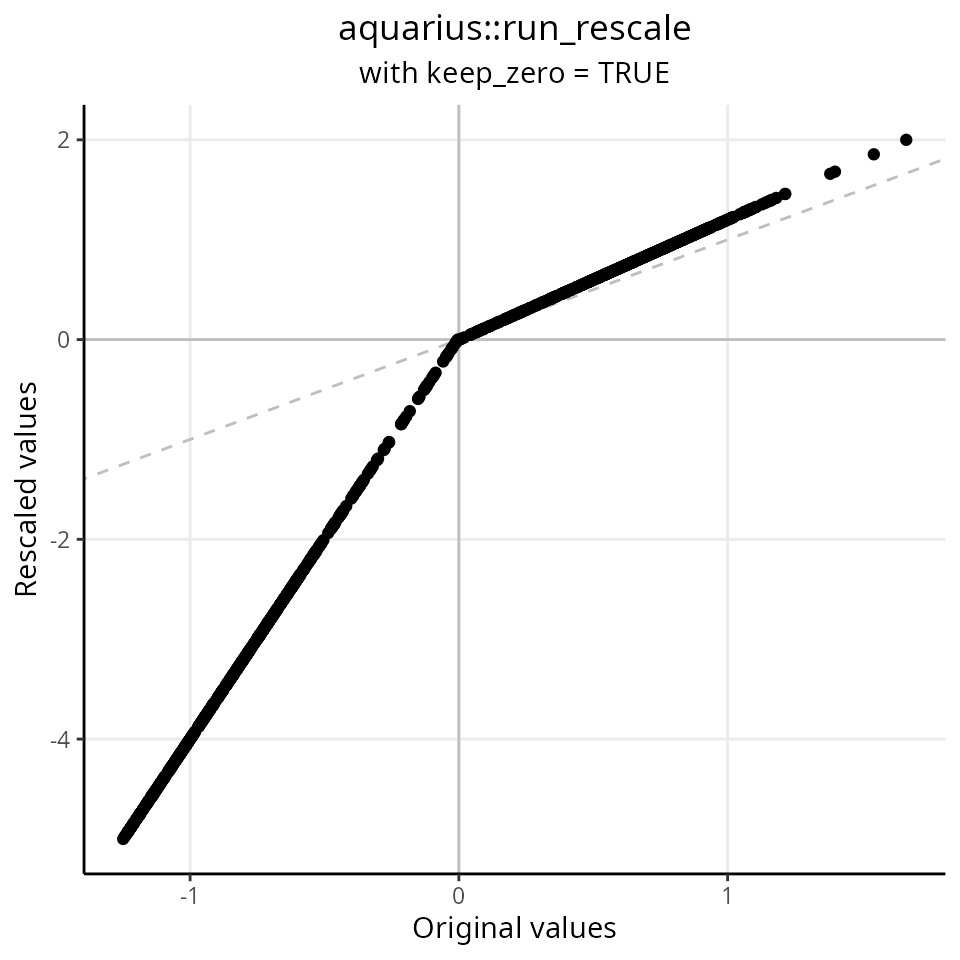
sample_cell_barcode
We visualize the clustering:
Seurat::DimPlot(sobj,
reduction = "harmony_48_tsne",
group.by = "seurat_clusters",
label = TRUE) +
Seurat::NoAxes() + Seurat::NoLegend() +
ggplot2::theme(aspect.ratio = 1)
We subset a maximum of 10 cells per cluster:
Seurat::Idents(sobj) = sobj$seurat_clusters
subsobj = aquarius::sample_cell_barcode(sobj,
n = 10)
subsobj## An object of class Seurat
## 13774 features across 80 samples within 1 assay
## Active assay: RNA (13774 features, 2000 variable features)
## 3 layers present: scale.data, data, counts
## 6 dimensional reductions calculated: RNA_pca, RNA_pca_48_tsne, RNA_pca_48_umap, harmony, harmony_48_umap, harmony_48_tsneWe visualize the projection with the remaining cells:
Seurat::DimPlot(subsobj,
reduction = "harmony_48_tsne",
group.by = "seurat_clusters",
label = TRUE) +
Seurat::NoAxes() + Seurat::NoLegend() +
ggplot2::theme(aspect.ratio = 1)
Cleaning :)
rm(subsobj)Functions to add
To be added in this vignette:
plot_externalread_gtfprepare_gsearepro_dependency_treerepro_installation_order
R Session
show
## R version 4.4.3 (2025-02-28)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.10
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.12.0
## LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.12.0
##
## locale:
## [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
## [3] LC_TIME=fr_FR.UTF-8 LC_COLLATE=en_US.UTF-8
## [5] LC_MONETARY=fr_FR.UTF-8 LC_MESSAGES=en_US.UTF-8
## [7] LC_PAPER=fr_FR.UTF-8 LC_NAME=C
## [9] LC_ADDRESS=C LC_TELEPHONE=C
## [11] LC_MEASUREMENT=fr_FR.UTF-8 LC_IDENTIFICATION=C
##
## time zone: Europe/Paris
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] ggplot2_3.5.2 patchwork_1.3.0 dplyr_1.1.4
##
## loaded via a namespace (and not attached):
## [1] RColorBrewer_1.1-3 shape_1.4.6.1
## [3] jsonlite_2.0.0 magrittr_2.0.3
## [5] spatstat.utils_3.1-3 farver_2.1.2
## [7] rmarkdown_2.29 zlibbioc_1.52.0
## [9] GlobalOptions_0.1.2 fs_1.6.6
## [11] ragg_1.4.0 vctrs_0.6.5
## [13] ROCR_1.0-11 spatstat.explore_3.4-2
## [15] S4Arrays_1.6.0 htmltools_0.5.8.1
## [17] SparseArray_1.6.2 sass_0.4.10
## [19] sctransform_0.4.2 parallelly_1.43.0
## [21] KernSmooth_2.23-26 bslib_0.9.0
## [23] htmlwidgets_1.6.4 desc_1.4.3
## [25] ica_1.0-3 plyr_1.8.9
## [27] plotly_4.10.4 zoo_1.8-14
## [29] cachem_1.1.0 igraph_2.1.4
## [31] mime_0.13 lifecycle_1.0.4
## [33] iterators_1.0.14 pkgconfig_2.0.3
## [35] Matrix_1.7-3 R6_2.6.1
## [37] fastmap_1.2.0 GenomeInfoDbData_1.2.13
## [39] MatrixGenerics_1.18.1 clue_0.3-66
## [41] fitdistrplus_1.2-2 future_1.40.0
## [43] shiny_1.10.0 digest_0.6.37
## [45] colorspace_2.1-1 S4Vectors_0.44.0
## [47] Seurat_5.3.0 tensor_1.5
## [49] RSpectra_0.16-2 irlba_2.3.5.1
## [51] GenomicRanges_1.58.0 textshaping_1.0.1
## [53] labeling_0.4.3 progressr_0.15.1
## [55] spatstat.sparse_3.1-0 httr_1.4.7
## [57] polyclip_1.10-7 abind_1.4-8
## [59] compiler_4.4.3 withr_3.0.2
## [61] doParallel_1.0.17 BiocParallel_1.40.2
## [63] fastDummies_1.7.5 MASS_7.3-65
## [65] DelayedArray_0.32.0 rjson_0.2.23
## [67] tools_4.4.3 lmtest_0.9-40
## [69] httpuv_1.6.16 future.apply_1.11.3
## [71] goftest_1.2-3 glue_1.8.0
## [73] nlme_3.1-168 promises_1.3.2
## [75] grid_4.4.3 Rtsne_0.17
## [77] cluster_2.1.8.1 reshape2_1.4.4
## [79] generics_0.1.3 gtable_0.3.6
## [81] spatstat.data_3.1-6 tidyr_1.3.1
## [83] data.table_1.17.0 XVector_0.46.0
## [85] sp_2.2-0 BiocGenerics_0.52.0
## [87] spatstat.geom_3.3-6 RcppAnnoy_0.0.22
## [89] ggrepel_0.9.6 RANN_2.6.2
## [91] foreach_1.5.2 pillar_1.10.2
## [93] stringr_1.5.1 spam_2.11-1
## [95] RcppHNSW_0.6.0 later_1.4.2
## [97] circlize_0.4.16 splines_4.4.3
## [99] lattice_0.22-6 survival_3.8-3
## [101] deldir_2.0-4 aquarius_1.0.0
## [103] tidyselect_1.2.1 SingleCellExperiment_1.28.1
## [105] ComplexHeatmap_2.23.1 miniUI_0.1.2
## [107] pbapply_1.7-2 knitr_1.50
## [109] gridExtra_2.3 IRanges_2.40.1
## [111] SummarizedExperiment_1.36.0 scattermore_1.2
## [113] stats4_4.4.3 xfun_0.52
## [115] Biobase_2.66.0 matrixStats_1.5.0
## [117] UCSC.utils_1.2.0 stringi_1.8.7
## [119] lazyeval_0.2.2 yaml_2.3.10
## [121] evaluate_1.0.3 codetools_0.2-20
## [123] tibble_3.2.1 cli_3.6.5
## [125] uwot_0.2.3 xtable_1.8-4
## [127] reticulate_1.42.0 systemfonts_1.2.3
## [129] jquerylib_0.1.4 GenomeInfoDb_1.42.3
## [131] Rcpp_1.0.14 globals_0.17.0
## [133] spatstat.random_3.3-3 png_0.1-8
## [135] spatstat.univar_3.1-2 parallel_4.4.3
## [137] pkgdown_2.1.2 ggalluvial_0.12.5
## [139] dotCall64_1.2 listenv_0.9.1
## [141] viridisLite_0.4.2 scales_1.4.0
## [143] ggridges_0.5.6 SeuratObject_5.1.0
## [145] purrr_1.0.4 crayon_1.5.3
## [147] GetoptLong_1.0.5 rlang_1.1.6
## [149] cowplot_1.1.3